Администрация городского округа Октябрьск Владельцам сельскохозяйственных животных
На территории Сызранского района выявлено заболевание крупного рогатого скота туберкулезом.
Далеко не все знают, что заболеванию туберкулезом подвержен не только человек, но и большинство домашних животных. К нему восприимчивы крупный рогатый скот, свиньи, куры, реже — кошки, собаки, гуси и утки, и лишь в виде исключения — овцы, лошади и ослы.
Причиной заболевания являются микобактерии Mycobacterium tuberculosis, именуемые ещё палочками Коха. Они очень устойчивы и могут находиться и жить в воде и почве до 4-х лет.
Источником заражения является заболевшее животное и его выделения — мокроты, испражнения, молоко. Пути заражения — воздушно-капельным путём или через пищу и питьё. Распространению заболевания способствует скученность животных, общий выпас и совместное поение и кормление.
Телята могут заразиться от больной матери внутриутробно.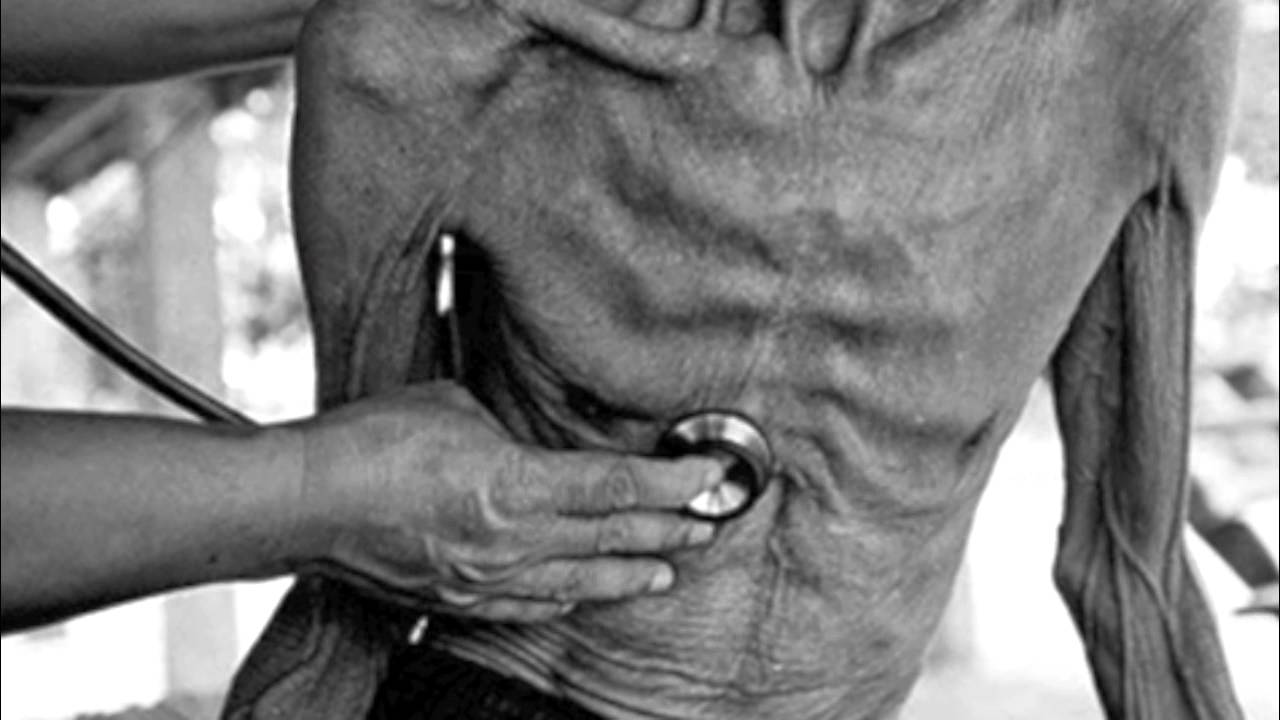 Не исключены случаи и передачи туберкулеза при контакте с больными людьми, которые осуществляют уход за животными.
Не исключены случаи и передачи туберкулеза при контакте с больными людьми, которые осуществляют уход за животными.
Поражение свиней возможно в процессе скармливания кухонных отходов, полученных в больницах и туберкулезных диспансерах и не подвергнутых обеззараживанию. Путь заражения кошек и собак — при питании молоком или мясом от больных коров.
Самые частые проявления болезни у животных:
- Повышение температуры носит постоянный характер.
- Потеря веса и аппетита.
- Одышка.
- Кашель с мокротой. В слизи может находиться кровь и некротические ткани.
- Сильное распухание заглоточных и подчелюстных лимфатических узлов на шее. Может сопровождаться выделением слюны и удушьем.
- Если поражено вымя, то при доении коровы в молоке могут быть кровь и творожистые массы. Лимфатические узлы возле вымени увеличиваются, становятся плотными и бугристыми.
- При кишечной форме туберкулеза наблюдается зловонный понос, с кровянистыми или гнойными примесями.
 Поносы чередуются с запорами и кишечными коликами, которые причиняют животному дискомфорт. Истощение организма животного при кишечной форме заболевания носит стремительный характер из — за того, что пища не усваивается.
Поносы чередуются с запорами и кишечными коликами, которые причиняют животному дискомфорт. Истощение организма животного при кишечной форме заболевания носит стремительный характер из — за того, что пища не усваивается. - Если у животного генерализованная форма заболевания, то ко всем перечисленным выше симптомам добавляются увеличенные лимфатические узлы по всему телу.
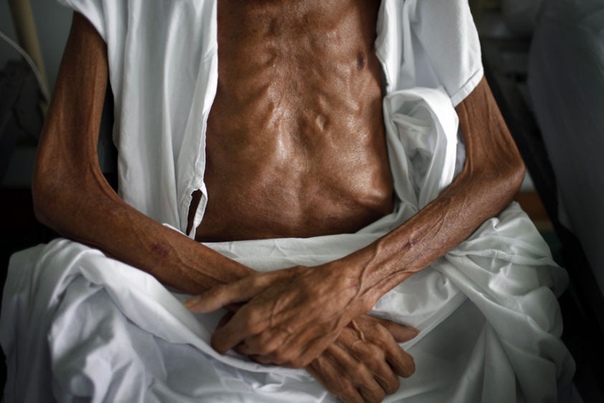 Поносы чередуются с запорами и кишечными коликами, которые причиняют животному дискомфорт. Истощение организма животного при кишечной форме заболевания носит стремительный характер из — за того, что пища не усваивается.
Поносы чередуются с запорами и кишечными коликами, которые причиняют животному дискомфорт. Истощение организма животного при кишечной форме заболевания носит стремительный характер из — за того, что пища не усваивается.
Туберкулез у животных не лечится, он может принести большой ущерб хозяйству, содержащему крупный рогатый скот. Поэтому необходимо проводить вакцинацию скота, регулярно делать туберкулиновые пробы (крупный рогатый скот исследуют два раза в год: весной, перед выгоном на пастбище, и осенью, перед постановкой скота на зимнее содержание, а молодняк крупного рогатого скота с 2-месячного возраста, и принимать следующие профилактические меры:
- Соблюдать гигиенические нормы, санитарные и ветеринарные правила при содержании, кормлении, транспортировке скота.
- Обеспечить 100% идентификацию.
- Регистрировать всех приобретенных животных в государственной ветеринарной службе и местной администрации.
- В течение 30 дней обеспечить вновь приобретенному скоту карантин.
- Закупку кормов проводить только у надёжных производителей с ветеринарными сопроводительными документами.
- Не допускать к работе на ферме работников без прохождения флюорографии или при наличии у них положительной реакции на заболевание.
- Обследоваться самостоятельно на наличие возбудителей туберкулёза в организме.
- При малейших признаках любого заболевания обращаться к ветеринару.
- Любое перемещение КРС, продажу молочной и мясной продукции, приобретение, транспортировку проводить исключительно после получения разрешения в государственной ветеринарной службе.
- Соблюдение всех рекомендаций и ограничений ветеринарной службы при выявлении туберкулёзной палочки в хозяйстве.
- В течение 14 дней сдавать на убой больных животных при выявлении заболевания.
Также важным профилактическим моментом выступает периодическая дезинфекция помещений, в которых содержаться коровы, чистка кормушек, поилок и прочего инвентаря, борьба с вредителями (грызунами, клещами и прочими). Чистый коровник — лучшая профилактика!
Можно ли пить молоко и есть мясо больных животных? Возбудитель туберкулёза может просуществовать в молоке 10 дней, в кисломолочной среде (кефир, ряженка, простокваша) может продержаться до 3-х недель. В сырах и сливочном масле палочка туберкулёза может быть активной до года, а мороженое может обеспечить существование инфекции до 6,5 лет. Молоко и мясо, заражённые туберкулёзной палочкой, представляют угрозу для здоровья человека, и употреблять их в пищу строго запрещено.
Важно! Чтобы избежать попадания туберкулёзной палочки в организм, приобретайте молочную продукцию у сертифицированных продавцов в специализированных магазинах, а не на стихийных рынках. Молоко сырое перед употреблением необходимо прокипятить.
Туберкулез может стать причиной потери всего поголовья фермерского хозяйства, тем самым сведя на нет весь многолетний труд его владельца. Кроме того, от инфицированных животных вполне может заразиться и человек. Именно поэтому следует со всей ответственностью подойти к соблюдению профилактических мер на ферме, а при обнаружении даже малейших признаков болезни, немедленно обращаться за помощью в ветеринарную службу.

Государственная ветеринарная служба
Сызранского района и
городского округа Октябрьск
О туберкулезе — БУ «Нижневартовская городская поликлиника»
Туберкулез – широко распространенное инфекционное заболевание, представляющее угрозу для жизни и здоровья окружающих. Туберкулез может поражать практически любые органы человека, но наибольшую опасность не только для больного, но и для окружающих представляет туберкулез органов дыхания. Ежегодно туберкулез уносит 2 миллиона жизни людей, при этом вновь заболевают 8 миллионов человек, а инфицировано микобактериями туберкулеза свыше 2 миллиарда человек, т. е. треть населения планеты.
е. треть населения планеты.
Смертность от туберкулеза в РФ продолжает снижаться, сейчас она составляет около 7%. При этом, по данным ВОЗ, туберкулез продолжает оставаться одной из 10 основных причин смертности в мире, опережая ВИЧ и малярию.
Болезнь глубоко укоренилась в группах населения с ущемленными правами человека и достоинства, — говорится в сообщении на сайте ВОЗ.
Характерной чертой современного течения туберкулеза является его сочетание с другими тяжелыми и социально значимыми инфекциями (ВИЧ, сифилис, гепатиты В и С).
Особенностью современного туберкулеза является увеличение лекарственной устойчивости микобактерий туберкулеза.
Возбудителя туберкулеза открыл в 1882 г. Р.Кох. Микобактерии туберкулеза обладают большой устойчивостью к неблагоприятным воздействиям внешних физических и химических факторов вследствие особенностей клеточной стенки и высокого содержания в ней липидов.
МБТ в естественных условиях при отсутствии солнечного света сохраняют свою жизнеспособность в течение нескольких месяцев, при рассеянном свете микобактерии погибают через 1-1,5 мес.
В уличной пыли возбудители туберкулеза сохраняются до 10 сут., на страницах книг – до 3 месяцев, в воде – до 5 мес. В то же время облученная солнечным светом культура МБТ погибает через 1,5 часа, а под воздействием ультрафиолетовых лучей – через 2 -3 минуты.
МБТ выдерживают нагревание при 70 градусов в течение 7 часов. При кипячении влажной мокроты микобактерии погибают через 5 мин., а высушенной мокроты – через 25 мин.
Пути передачи туберкулезной инфекции: аэрогенный путь заражения является ведущим. Это происходит воздушно – капельным или воздушно – пылевым путем. При кашле, чихании или разговоре микобактерии с капельками слизи или мокроты попадают во внешнюю среду. Важен и пищевой путь передачи, который реализуется при употреблении инфицированных продуктов животного происхождения (молоко, сметана, сыр, творог).
Диагностика туберкулеза включает обязательный диагностический минимум: сбор жалоб и анамнеза, объективное исследование больного, анализы крови и мочи, рентгенографию грудной клетки или флюорографию, микроскопическое исследование мокроты не менее 3 раз, а также диаскинтест, разработанный учеными из Московской академии им.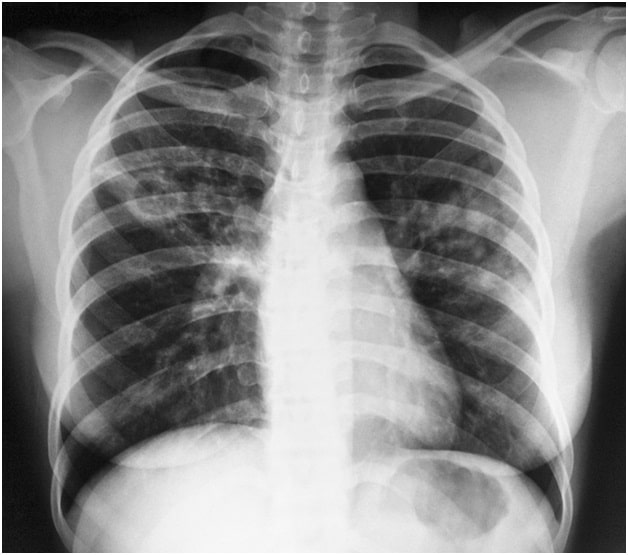 Сеченова. Это внутрикожный диагностический тест повышенной восприимчивости к туберкулезу, который стал альтернативой туберкулиновому тесту. Последний часто давал ошибочные положительные реакции на микобактерии туберкулеза у здоровых и вакцинированных БЦЖ людей.
Сеченова. Это внутрикожный диагностический тест повышенной восприимчивости к туберкулезу, который стал альтернативой туберкулиновому тесту. Последний часто давал ошибочные положительные реакции на микобактерии туберкулеза у здоровых и вакцинированных БЦЖ людей.
Вторая перспективная разработка – это биочип, созданный в НИИ Энгельгарта. Биочип позволяет определить геномную структуру микобактерии туберкулеза, резистентную к противотуберкулезным препаратам.
Для человека МБТ – чужеродный организм. В норме при ее появлении и размножении в организме иммунные клетки атакуют возбудителя заболевания, не допуская стадии активного размножения. Развитие болезни возможно в двух случаях: 1. если иммунная система подавлена, есть нарушения выработки антител, состояния иммунодефицитов, защитные силы организма ослаблены иными заболеваниями; 2. или есть контакт с возбудителем длительный, постоянный, бациллоноситель находится на стадии открытой формы заболевания и не получает необходимого лечения (при недиагностированном туберкулезе у члена семьи, при содержании в закрытых учреждениях).
Среди факторов, снижающих специфический иммунитет и способствующих развитию туберкулеза при контакте с инфекционным агентом, выделяют следующие:
- табакокурение, как фактор развития заболеваний бронхолегочной системы, ослабляющий местный иммунитет;
- неумеренный прием алкогольных напитков;
- все виды наркоманий;
- предрасположенность к болезням органов дыхательной системы из –за наличия аномалий строения, частых заболеваний в анамнезе, наличия хронических воспалительных процессов в органах дыхания;
- хронические заболевания и очаги в других органах и тканях;
- сахарный диабет, эндокринные заболевания;
- несбалансированное питание, недостаточность витаминов, питательных веществ, ведь как известно, слово «фтизиатрия» – название раздела медицины, изучающего туберкулез, происходит от греческого «phtisis», что означает истощение;
- невротические нарушения, депрессивные состояния, низкая стрессоустойчивость;
- период беременности;
- неблагоприятные социально – бытовые условия.

Туберкулез излечим. В целях избежания данного недуга ведите здоровый образ жизни, занимайтесь умеренными физическими нагрузками, правильно питайтесь, делайте профилактические прививки, ежегодно проходите флюорографию органов дыхания.
Количество просмотров: 879
Туберкулез у детей
Актуально
16:32 26/03/2020
Просмотров: 970
Туберкулез – опасное инфекционное заболевание, которое вызывается микобактерией(бациллой Коха). Чаще всего оно поражает легкие, хотя в процесс могут вовлекаться и другие органы.
Туберкулез в мире остается одной из лидирующих причин смерти. В России в последние годы ситуация также ухудшается: растет количество заболевших, а значит и распространение палочки Коха среди населения. Будучи нераспознанным и нелеченным, туберкулез может иметь фатальные последствия. Однако предотвратить и вылечить его возможно.
Как происходит заражение туберкулезом
Заражение туберкулезом происходит через легкие от больного человека, который выделяет бациллы во внешнюю среду при кашле или чихании.![]() Палочка туберкулеза очень устойчива и может сохраняться в уличной пыли до 2 месяцев. Человек, вдохнувший зараженные частицы воздуха, становится контактным, а бацилла оседает у него в организме. Такой контактный носитель может быть среди вашего окружения, родственников и близких людей. По подсчетам ученых, до 1/3 населения Земли являются скрытыми носителями бациллы туберкулеза.
Палочка туберкулеза очень устойчива и может сохраняться в уличной пыли до 2 месяцев. Человек, вдохнувший зараженные частицы воздуха, становится контактным, а бацилла оседает у него в организме. Такой контактный носитель может быть среди вашего окружения, родственников и близких людей. По подсчетам ученых, до 1/3 населения Земли являются скрытыми носителями бациллы туберкулеза.
Несмотря на попадание инфекции, у большинства зараженных людей туберкулез не развивается. Только 5-10% носителей на протяжении последующей жизни заболевают активной формой туберкулеза, в то время как у остальных бацилла сохраняется в спящем состоянии под контролем иммунной системы.
Особенности детского туберкулеза
Для детского организма, особенно до 2 лет, туберкулезная палочка является намного более опасной, чем для взрослых. У детей процент перехода болезни в активную форму гораздо выше, а также очень высок риск развития тяжелых генерализованных форм: милиарного туберкулеза, сепсиса, туберкулезного менингита.
Большую роль в развитии туберкулеза играет не только сам факт попадания бактерии в организм, но и состояние инфицированного ребенка. Сильными предрасполагающими факторами являются плохое питание, авитаминоз, истощение, постоянный стресс и недосыпание – другими словами, неблагоприятные условия жизни. Вот почему дети из бедных и маргинальных семей наиболее подвержены риску заболеть.
Симптомы туберкулеза у детей
Чаще всего легочный туберкулез проявляется кашлем. В начале болезни он может напоминать бронхит или даже простуду. Однако, вместо того, чтобы поправиться через неделю, ребенок продолжает болеть, кашель усиливается, мокрота может приобретать розовый оттенок. Ребенок выглядит истощенным, худеет, теряет вес. Температура повышается к вечеру, а днем может быть нормальной.
Поэтому при любых длительных заболеваниях легких врач должен направить ребенка на обследование в противотуберкулезый диспансер (ПТД). В обследование входят тщательный осмотр, рентген легких, подробные анализы крови, посев мокроты, может потребоваться компьютерная томография. Не стоит отказываться от похода в ПТД, если вас направил участковый педиатр – здоровье ребенка дороже предрассудков.
Не стоит отказываться от похода в ПТД, если вас направил участковый педиатр – здоровье ребенка дороже предрассудков.
Внелегочный туберкулез (костный, суставной, кожный и так далее) проявляется по-разному, в зависимости от места внедрения возбудителя. Непременным симптомом является повышенная температура и увеличение местных лимфоузлов.
Важным диагностическим критерием туберкулеза служит реакция Манту. Она показывает, встречался ли ребенок с туберкулезной палочкой ранее. Диагностическая ценность этого метода далека от 100%, случаются ложноположительные и трудно интерпретируемые результаты. Вместо Манту в наше время есть более современный способ – внутрикожная проба с препаратом Диаскинтест.
Профилактика туберкулеза у детей
В нашей стране в связи с широким распространением туберкулеза всем детям в роддоме вводится вакцина БЦЖ или БЦЖ-М (в зависимости от того, есть ли в семье ребенка взрослые, способные выделять палочки туберкулеза). Это является необходимой мерой первичной профилактики, которая значительно снижает риск тяжелых форм туберкулеза (менингит, сепсис) и уменьшает смертность от этой инфекции.
Однако БЦЖ не является панацеей, и маленького ребенка во избежание заражения нужно всячески ограждать от контакта с потенциально больными людьми: по возможности не пользоваться общественным транспортом, избегать вокзалов и других мест общественного пользования, большого скопления людей. Если в семье имеется родственник, больной активной формой туберкулеза, ребенок не должен жить в одной с ним квартире и общаться до полного прекращения выделения им туберкулезной палочки.
Детям,ранее не инфицированным, в возрасте 6-7 лет проводится ревакцинация БЦЖ, поскольку иммунитет со временем угасает. Для выявления групп риска детям в возрасте с 1года до 7 лет проводится проба Манту, детям и подросткам (8-17 лет) проводится проба Диаскинтест.
ТУБЕРКУЛЕЗ ЖИВОТНЫХ — СОЦИАЛЬНАЯ БОЛЕЗНЬ! КЛИНИЧЕСКИЕ ПРИЗНАКИ, ПРОФИЛАКТИКА.
Туберкулёз – социальная болезнь!
Туберкулёз – хроническое инфекционное заболевание практически всех видов животных и человека, характеризующееся образованием в различных органах специфических бугорков – туберкулов, вызываемая патогенными микобактериями туберкулёза.
Туберкулёз крупного рогатого скота наносит значительный экономический ущерб животноводству и представляет опасность заражения людей. Поэтому организация и проведение обязательных противотуберкулезных мероприятий были и остаются актуальной проблемой.
За последние 12 лет на территории Калужской области было установлены два неблагополучных пункта по туберкулёзу крупного рогатого скота (в 2007 году на территории Юхновского района и в 2019 году на территории Хвастовичского района).
С учётом, что Калужская область в целом на протяжении 20 лет благополучна по туберкулёзу, был принят метод оздоровления хозяйств путём полной единовременной замены поголовья животных. Необходимо отметить, что любой метод оздоровления хозяйства несёт большие экономические затраты.
Туберкулёз занимает особое место среди инфекционных болезней животных. Он своеобразен тем, что долгие годы может протекать в скрытой форме, без проявления клинических признаков болезни, не влияя на продуктивность и жизнедеятельность животных. Туберкулёз легко и быстро распространяется среди поголовья крупного рогатого скота, но искореняется с большим трудом. В неблагополучных стадах достаточно остаться одному не выявленному больному животному, чтобы на долгие годы растянуть проведение оздоровительных мероприятий. Резервуар инфекции так разнообразен и обширен, что мероприятия по борьбе с туберкулёзом крупного рогатого скота не могут ограничиться только этим видом животных, а должны обхватить всех домашних и диких животных, а также объекты внешней среды.
Туберкулёз легко и быстро распространяется среди поголовья крупного рогатого скота, но искореняется с большим трудом. В неблагополучных стадах достаточно остаться одному не выявленному больному животному, чтобы на долгие годы растянуть проведение оздоровительных мероприятий. Резервуар инфекции так разнообразен и обширен, что мероприятия по борьбе с туберкулёзом крупного рогатого скота не могут ограничиться только этим видом животных, а должны обхватить всех домашних и диких животных, а также объекты внешней среды.
Большинство исследователей считает, что каждый вид возбудителя туберкулёза обладает патогенностью преимущественно для определённого вида животных. Так, возбудитель туберкулёза бычьего вида в основном патогенен для крупного рогатого скота, возбудитель человеческого вида – для человека, возбудитель птичьего вида – для птиц. Однако те или иные виды микобактерий выделяются из организма различных видов животных, способны циркулировать среди животных и человека, но патогенность этих микобактерий неодинакова для разных видов животных. Так, микобактерии туберкулёза бычьего вида, кроме крупного рогатого скота, патогенны для свиней, коз, овец, лошадей, верблюдов, собак, кошек, маралов, кроликов, морских свиной и других животных, а также человека. Микобактерии туберкулёза человеческого вида, кроме человека, патогенны для свиней, собак, кошек, обезьян. Микобактерии птичьего вида, кроме птиц, патогенны для кроликов и свиней. Некоторые виды животных устойчивы, а другие чувствительны к двум или ко всем трём видам микобактерий туберкулёза, что зависит от естественной резистентности и реактивности животных и человека, от возможного контакта и факторов передачи возбудителя.
Так, микобактерии туберкулёза бычьего вида, кроме крупного рогатого скота, патогенны для свиней, коз, овец, лошадей, верблюдов, собак, кошек, маралов, кроликов, морских свиной и других животных, а также человека. Микобактерии туберкулёза человеческого вида, кроме человека, патогенны для свиней, собак, кошек, обезьян. Микобактерии птичьего вида, кроме птиц, патогенны для кроликов и свиней. Некоторые виды животных устойчивы, а другие чувствительны к двум или ко всем трём видам микобактерий туберкулёза, что зависит от естественной резистентности и реактивности животных и человека, от возможного контакта и факторов передачи возбудителя.
Туберкулёз зарегистрирован почти у всех млекопитающих, птиц, рыб и хладнокровных. Эпизоотический и эпидемический процесс туберкулёза состоит из трёх составных частей: источника инфекции, механизма передачи возбудителя и восприимчивых к ней животных и людей.
Процесс распространения туберкулёза среди животных принято называть эпизоотическим процессом туберкулёза. Это процесс представляет собой цепь последовательных заражений одного животного от другого зараженного туберкулёзом животного. Возбудитель туберкулёза циркулирует по нескольким вариантам: — животное – человек, человек – человек, человек – животное, животное – животное.
Это процесс представляет собой цепь последовательных заражений одного животного от другого зараженного туберкулёзом животного. Возбудитель туберкулёза циркулирует по нескольким вариантам: — животное – человек, человек – человек, человек – животное, животное – животное.
Первичным источником инфекции при туберкулёзе крупного рогатого скота является зараженный организм животного, который является естественной средой обитания, сохранения, размножения и накопления патогенного возбудителя.
Таким образом, в настоящее время риск заражения животных от больных туберкулёзом людей заметно повысился и продолжает повышаться. В последние годы всё чаще появляются сообщения о том, что эпизоотическая обстановка по туберкулёзу начинает сказываться на росте заболеваемости людей.
В целях профилактики туберкулёза местные (районные) центры санитарно-эпидемиологического надзора и участковые врачи сельских (поселковых) поликлиник (амбулаторий) не должны допускать к работе в животноводстве и кормопроизводстве лиц, не прошедших обследование на туберкулез, а также больных туберкулезом и находящихся в группе диспансерного учета, обязаны организовать постоянное медицинское наблюдение за персоналом, обслуживающим животных, установить контроль за обеспечением их спецодеждой и спецобувью, умывальниками, мылом, полотенцами и средствами для дезинфекции рук и обуви; в случае установления заболевания обслуживающего персонала туберкулезом больных людей немедленно освободить от работы по обслуживанию животных; совместно со специалистами государственной ветеринарной сети и хозяйств установить контроль за проведением обязательной пастеризации молока и термической обработки других сырых молочных продуктов на неблагополучных фермах хозяйств, молокозаводах и других предприятиях по переработке молока и молочных продуктов.
При естественном заражении возбудителем туберкулёза инкубационный период в большинстве случаев установить не представляется возможным. Экспериментально определено, что от момента заражения до появления положительной реакции на туберкулин проходит не менее 15 дней. Клинические признаки могут появиться через месяцы и даже через годы после заражения. Ухудшение общего состояния животного при туберкулёзе выражается в прогрессирующем исхудании при сохранении аппетита и удовлетворительном кормлении, быстрой утомляемости при передвижении и снижении продуктивности. Температура тела временами повышается, более стойкое повышение температуры тела наблюдается при различного рода обострениях течения процесса туберкулёза.
Клиническое проявление туберкулёза у крупного рогатого скота зависит от локализации процесса и степени поражения органов. Проявление клинических признаков можно было наблюдать у больных животных, которых годами содержали в туберкулёзных изоляторах. В последние годы клинически больные животные практически не встречаются, так как ежегодно во всех хозяйствах после каждой туберкулинизации реагирующий скот сдают на убой в течении 15 дней после исследований.
Начальные стадии туберкулёза, как правило, не имеют клинических признаков. Заметными становятся лишь значительные поражения, сопровождающиеся нарушением общего состояния организма или расстройством функций отдельных органов.
Клинические признаки туберкулёза у крупного рогатого скота не являются типичными только для этого заболевания, и часто они вообще отсутствуют. Тем не менее, клиническое обследование проводить необходимо, поскольку некоторые больные животные, особенно с пониженной упитанностью, утрачивают способность реагировать на туберкулин. Одновременно с этим они могут иметь открытый туберкулёзный процесс и являться очень опасным источником распространения возбудителя туберкулёза. Такие клинические признаки, как истощение животных при нормальном кормлении, кашель, особенно по утрам, когда открываются двери коровника, учащенное дыхание, периодическое появление лихорадки, увеличение и бугристость наружных (подчелюстных, предлопаточных, надвымянных) лимфатических узлов, должны вызывать подозрение на наличие туберкулёза. В длительно неблагополучных по туберкулёзу пунктах симптомы поражения кишечника туберкулёзом могут выражаться в форме расстройства пищеварения, поносов, чередующихся с запорами. Туберкулёз остаётся одним из широко распространённых, древнейших и сложных инфекционный заболеваний человека и животных, для которого до настоящего времени не разработаны высокоэффективные средства иммунной защиты и лечения. Поэтому в настоящее время основой профилактических и оздоровительных мероприятий при туберкулёзе животных была и остаётся диагностика болезни. Все мероприятия по профилактике туберкулёза, в том числе диагностике и по ликвидации в случае возникновения заболевания проводятся в соответствии с «Профилактика и борьба с заразными болезнями, общими для человека и животных. Туберкулез. Санитарные правила. Ветеринарные правила. СП 3.1.093-96, ВП 13.3.1325-96».
В длительно неблагополучных по туберкулёзу пунктах симптомы поражения кишечника туберкулёзом могут выражаться в форме расстройства пищеварения, поносов, чередующихся с запорами. Туберкулёз остаётся одним из широко распространённых, древнейших и сложных инфекционный заболеваний человека и животных, для которого до настоящего времени не разработаны высокоэффективные средства иммунной защиты и лечения. Поэтому в настоящее время основой профилактических и оздоровительных мероприятий при туберкулёзе животных была и остаётся диагностика болезни. Все мероприятия по профилактике туберкулёза, в том числе диагностике и по ликвидации в случае возникновения заболевания проводятся в соответствии с «Профилактика и борьба с заразными болезнями, общими для человека и животных. Туберкулез. Санитарные правила. Ветеринарные правила. СП 3.1.093-96, ВП 13.3.1325-96».
Особенности питания при туберкулезе — БУ «Республиканский противотуберкулезный диспансер» Минздрава Чувашии
Особенности питания при туберкулезе
Мотивирование граждан к ведению здорового образа жизни посредством информационно-коммуникационной кампании — одна из целей регионального проекта «Формирование системы мотивации граждан к здоровому образу жизни, включая здоровое питание и отказ от вредных привычек»
О том, как правильно питаться при заболевании туберкулезом рассказывают специалисты отделения дневного стационара Республиканского противотубркулезного диспансера Минздрава Чувашии.
Туберкулез легких – инфекционное заболевание, причиной которого является туберкулезная палочка – агрессивный и устойчивый микроб. Правильное питание при данном заболевании способно снизить интоксикацию организма, а также повысить сопротивляемость к болезни. Питание больного при туберкулезе должно быть сбалансированным и с высоким содержанием белка, так как в организме больного белки распадаются быстрее, чем у здорового человека.
Меню должно включать молочные продукты, яйца, домашнюю птицу, рыбу телятину.
В рационе обязательно должны быть фрукты и овощи, богатые витаминами С, В, А: киви, лимон, апельсины, капуста, болгарский перец.
Необходимы и продукты, содержащие жиры: сливочное масло, растительные масла (подсолнечное, оливковое), рыбий жир. Однако не стоит забывать, что избыточное содержание жиров в рационе может привести к расстройству пищеварения, болезням печени.
Также в рационе должны быть продукты, богатые углеводами: каши, различные мучные изделия, хлеб, мед.![]()
Диета при истощении и туберкулезе легких разрешает употреблять любые продукты питания, однако, следует убрать из рациона очень жирные сорта рыбы и птицы, бараний, говяжий и кулинарные жиры. Кроме того, стоит отказаться от острых и жирных соусов, тортов и пирожных с большим содержанием крема.
Питание больного при туберкулезе легких, в первую очередь, должно быть калорийным, но это вовсе не значит, что нужно стремиться к перекармливанию больного.
У большинства больных туберкулезом практически полностью отсутствует аппетит, а правильный режим питания при данном заболевании требует принятия пищи не менее 4-5- раз в день. Таким образом, основный вывод по поводу питания при туберкулезе легких: пища должна быть максимально здоровой и иметь аппетитный вид и запах, приготовлена исключительно из свежих продуктов.
(PDF) Investigation of nutritional status and body composition in patients with tuberculosis
Руднев С.Г. и др. Исследование нутритивного статуса и состава тела больных туберкулезом
106
Пульмонология 1’2013
поддержки в системе противотуберкулезных меро$
приятий необходимы дальнейшие исследования.
Простым и надежным методом определения со$
става тела, пригодным для мониторинга эффектив$
ности диетотерапии в комплексном лечении больных
туберкулезом, является биоимпедансный анализ. По$
мимо характеристики состава тела биоимпедансный
анализ позволяет определять энергетические по$
требности организма, что создает основу для индиви$
дуализации режимов диетотерапии. Другая потенци$
альная возможность применения биоимпедансного
анализа во фтизиатрии связана с прогнозированием
исходов оперативного лечения на основе таких ин$
дикаторов, как индекс безжировой массы и фазовый
угол импеданса [35, 55].
Литература
1. Перельман М.И., Богадельникова И.В. Фтизиатрия. М.:
ГЭОТАР$Медиа; 2010.
2. Шилова М.В. Туберкулез в России в 2010 году. М.; 2012.
3. Диетическая терапия больных туберкулезом: Методи$
ческие рекомендации / Тутельян В.А., Каганов Б.С.,
Погожева А.В. и др. М.: Министерство здравоохране$
ния и социального развития РФ; 2010.
4. Уголев А.М. Теория адекватного питания и трофология.
СПб.: Наука; 1991.
5. Webster’s online dictionary, http://www.websters2online2dic2
tionary.org/definitions/ Nutritional+Status
6. Лейдерман И.Н., Николенко А.Г., Сивков О.Г. Нутритив$
ная поддержка в отделении реанимации и интенсив$
ной терапии. Стандартные алгоритмы и протоколы:
Учеб.$метод. пособие. М.; 2010.
7. Sobotka L., ed. Basics in clinical nutrition. 4th ed. Prague:
Galen; 2011.
8. Schindler K., Pernicka E., Laviano A. et al. How nutritional
risk is assessed and managed in European hospitals: a survey
of 21007 patients findings from the 2007–2008 cross$section$
al nutritionDay survey. Clin. Nutr. 2010; 29 (5): 552–559.
9. Аксенова В.А., Бирон М.Г., Корнилова З.Х. и др. Клини$
ческое значение лечебно$диетической коррекции бел$
ково$энергетической недостаточности у детей, подро$
стков и взрослых, больных туберкулезом органов дыха$
ния. Пульмонология 2010; 3: 73–78.
Пульмонология 2010; 3: 73–78.
10. Bernabe2Ortiz A., Carmano C.P., Sanchez J.F., Rios J. Weight
variation over time and its association with tuberculosis
treatment outcome: a longitudinal analysis. PloS One 2011;
6 (4): e18474.
11. Quetelet A. Physique sociale: ou essay sur le development des
faculties de l’homme. Brussels: C. Muquardt; 1869.
12. Keys A., Fidanza F., Karvonen M.J. et al. Indices of relative
weight and obesity. J. Chron. Dis. 1972; 25 (6–7): 329–342.
13. Calle E.E., Thun M.J., Petrelli J.M. et al. Body$mass index
and mortality in a prospective cohort of U.S. adults.
N. Engl. J. Med. 1999; 341 (15): 1097–1105.
14. Lonnroth K., Williams B.G., Cegielski P., Dye C. A consistent
log$linear relationship between tuberculosis incidence and
body mass index. Int. J. Epidemiol. 2010; 39 (1): 149–155.
15. Коновалова М.В., Анисимова А.В., Вашура А.Ю. и др.
Нутритивный статус детей с онкологическими заболе$
ваниями в состоянии ремиссии по данным биоимпеда$
нсного исследования. Онкогематология 2012; 2: 42–50.
Онкогематология 2012; 2: 42–50.
16. Каминская Г.О., Абдуллаев Р.Ю., Комиссарова О.Г.
Оценка нутритивного статуса больных туберкулезом
методами биохимического исследования. Туб. и бол.
легких 2011; 88: 178.
17. Титюхина М.В., Батыров Ф.А., Корнилова З.Х. Коррек$
ция метаболических нарушений у больных туберкуле$
зом легких с сопутствующими заболеваниями. Туб. и
бол. легких 2010; 87: 49–53.
18. Russell C.A., Elia M. Nutrition screening survey in the UK
in 2007. British Association of Parenteral and Enteral
Nutrition; 2008.
19. Bose K., Jana S., Bisai S. et al. Comparison of nutritional
status between tuberculosis patients and controls: a study
from North 24 Parganas district in West Benghal, India.
Mal. J. Nutr. 2007; 13 (2): 131–139.
20. Pakasi T.A., Karyadi E., Dolmans W.M. et al. Malnutrition
and socio$demographic factors associated with pulmonary
tuberculosis in Timor and Rote Islands, Indonesia. Int. J.
Int. J.
Tuberc. Lung Dis. 2009; 13 (6): 755–759.
21. Dodor E.A. Evaluation of nutritional status of new tubercu$
losis patients at the Effia$Nkwanta regional hospital. Ghana
Med. J. 2008; 42 (1): 22–28.
22. Zachariah R., Spielmann M.P., Harries A.D., Salaniponi F.M.L.
Moderate to severe malnutrition in patients with tuberculo$
sis is a risk factor associated with early death. Trans. Roy.
Soc. Trop. Med. Hyg. 2002; 96 (3): 291–294.
23. Miyata S., Tanaka M., Ihaku D. Subjective global assess$
ment in patients with pulmonary tuberculosis. Nutr. Clin.
Pract. 2011; 26 (1): 55–60.
24. Kim D.K., Kim H.J., Kwon S2Y. et al. Nutritional deficit as a
negative prognostic factor in patients with miliary tubercu$
losis. Eur. Respir. J. 2008; 32 (4): 1031–1036.
25. Mueller C., Compher C., Druyan M.E., ASPEN board of
directors. ASPEN clinical guidelines: nutrition screening,
assessment, and intervention in adults. J. Parent. Enter.
J. Parent. Enter.
Nutr. 2011; 35 (1): 16–24.
26. Soeters P.B., Reijven P.L.M., van Bokhorst2de van der
Schueren M.A.E. et al. A rational approach to nutritional
assessment. Clin. Nutr. 2008; 27 (5): 706–716.
27. Heymsfield S.B., Lohman T.G., Wang Z., Going S.B. Human
body composition. 2nd ed. Champaign, IL: Human Kinetics;
2005.
28. Мартиросов Э.Г., Николаев Д.В., Руднев С.Г. Техноло$
гии и методы определения состава тела человека. М.:
Наука; 2006.
29. Kershaw E.E., Flier J.S. Adipose tissue as an endocrine
organ. J. Clin. Endocrinol. 2004; 89 (6): 2548–2556.
30. Magnus2Levy A. Physiology des Stoffwechsels. In: Van Noor$
den C., ed. Handbuch der Pathologie des Stoffwechsels.
Berlin: Hirschwald; 1906.
31. Paton N.I., Ng Y.2M. Body composition studies in patients
with wasting associated with tuberculosis. Nutrition 2006;
22 (3): 245–251.
32. Brozek J., Wells S., Keys A. Medical aspects of semistarva$
tion in Leningrad (siege 1941–1942). Am. Rev. Soviet Med.
Am. Rev. Soviet Med.
1946; 4 (1): 70–86.
33. Schols A.M.W.J., Broekhuizen R., Weling2Scheepers C.A.,
Wouters E.F. Body composition and mortality in chronic
obstructive pulmonary disease. Am. J. Clin. Nutr. 2005; 82
(1): 53–59.
34. Schutz Y., Kyle U.U.G., Picard C. Fat$free mass index per$
centiles in Caucasians aged 18$98 y. Int. J. Obes. 2002; 26
(7): 953–960.
35. Slinde F., Gronberg A., Engstrom C.2P. et al. Body composi$
tion by bioelectrical impedance predicts mortality in chron$
ic obstructive pulmonary disease patients. Respir. Med.
2005; 99 (8): 1004–1009.
О возможной роли checkpoint-ингиборов в профилактике и лечении инфекционных заболеваний
В журнале «Nature Reviews Immunology» за
2017 год Michelle N. Wykes, Sharon
R.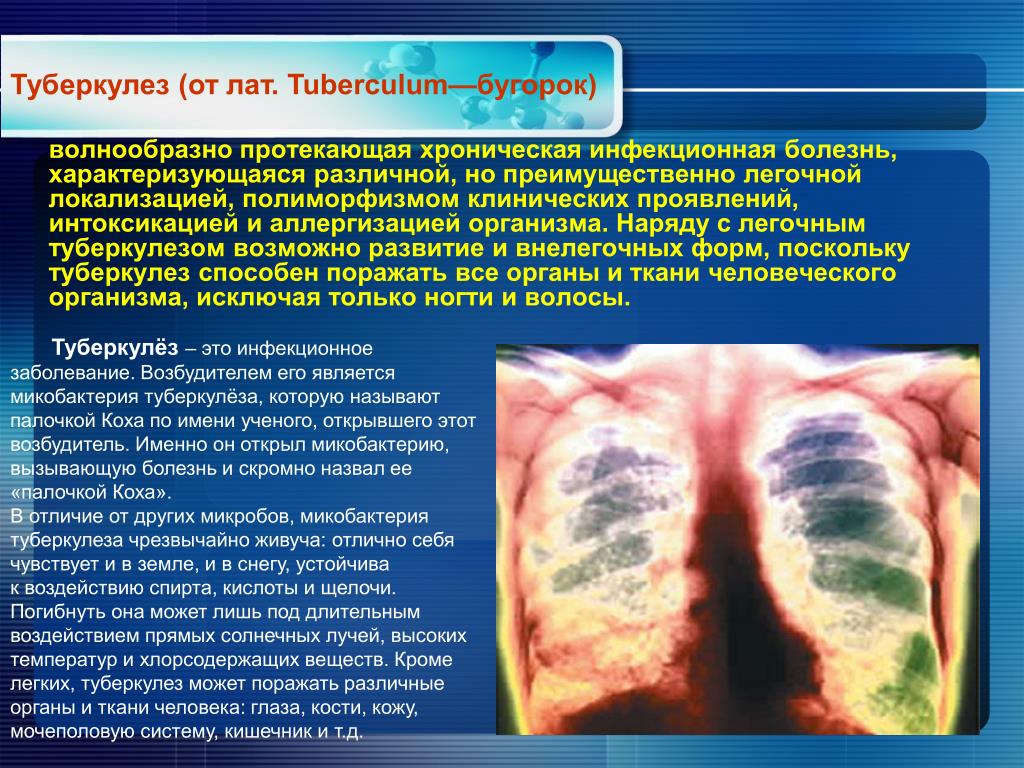 Lewin была представлена очень
Lewin была представлена очень
интересная статья, посвященная вопросу возможного применения checkpoint-ингибиторов
с целью профилактики и лечения целого ряда инфекционных заболеваний. В
данной статье описывается значение передачи сheckpoint-сигналов
в случае заражения малярией, ВИЧ и вирусом гепатита В, а также туберкулеза (ТБ), а также
возможности терапевтического воздействия на иммунные контрольные точки в условиях наличия
инфекционного заболевания.
При развитии острых инфекций, таких как малярия, а также при хронических вирусных
инфекциях, включая ВИЧ и вирус гепатита, наблюдается повышение активности точек иммунного
контроля — белка запрограммированной клеточной гибели 1 (PD1) и цитотоксического
Т-лимфоцитарного антигена 4 (CTLA4).
Успешное применение checkpoint-ингибиторов в терапии
злокачественных новообразований дает основание авторам статьи предполагать, что
использование препаратов данной группы также будет эффективным для профилактики и лечения
ряда социально-значимых инфекционных заболеваний.
Молекулы иммунной контрольной точки являются ингибиторными рецепторами, экспрессируемыми на
иммунных клетках, которые являются триггером иммуносупрессивных сигнальных путей. Опосредованная
передача сигналов через данные молекулы может привести к истощению иммунных клеток (особенно
Т-лимфоцитов). Истощение Т-клеток проявляется снижением их функции, устойчивой экспрессией
молекул иммунной контрольной точки (например, белка запрограммированной клеточной гибели 1
(PD1)), плохим обратным ответом и транскрипционным состоянием, отличным от состояния нормально
функционирующих эффекторных Т-лимфоцитов или Т-клеток памяти[3]. Существует множество типов
Существует множество типов
активирующих и ингибиторных взаимодействий, которые возникают между антиген-представляющими
клетками (АРС) и Т-клетками, и эти взаимодействия определяют характер иммунных реакций.
Рисунок 1 — Взаимодействие с антиген-представляющими
клетками, регулирующими ответы Т-клеток (адаптировано Michelle N. Wykes
et al., 2017).
Исследование иммуносупрессивных взаимодействий привело к клиническому развитию и внедрению новых
методов лечения рака, которые увеличивают возможности использования специфических антител в
качестве checkpoint-ингибиторов. Антитела, нацеленные на PD1 (Пембролизумаб, Ниволумаб), на
цитотоксический Т-лимфоцитарный антиген 4 (CTLA4) (Ипилимумаб) и лиганд программированной гибели
клеток 1 (PDL1) (Атезолизумаб, Авелумаб и Дурвалумаб), в настоящее время рекомендованы для
монотерапии различных видов злокачественных опухолей. Кроме того, было показано, что
Кроме того, было показано, что
комбинированная таргетная терапия, нацеленная на PD1 и CTLA4, является более эффективной, чем
любая другая терапия меланомы[4], хотя также имеет токсическое воздействие на организм.
Основная проблема иммунотерапии заключается в том, чтобы понять, почему существуют различные
ответы на лечение. На данный момент, идет поиск предиктивных «биомаркеров»
благоприятного клинического ответа. Например, выявление экспрессии PDL1 на опухолевых клетках
помогает в поиске тех пациентов, которые в наибольшей степени ответят на терапию, в которой
используется блокаторы PD1 или PDL1[5]. Учитывая стоимость и токсичность использования
checkpoint-ингибиторов, определение биомаркеров в настоящее время является главным приоритетом.
Эффективность иммунотерапии в лечении инфекционных заболеваний изучена не так хорошо. Однако,
Однако,
предполагается, что она может быть использована для профилактики и лечения инфекционных
заболеваний как в острой, так и в хронической фазах инфекции. Разработка вакцин для целого ряда
инфекционных заболеваний, включая малярию, вирус гепатита B (HBV) и ВИЧ, может быть улучшена за
счет использования checkpoint-ингибиторов. Учитывая, что резистентность к лекарственным
средствам, применяемых для лечения малярии[10] и многих других инфекциях возрастает, что ВИЧ и
HBV требуют лечения и контроля в течение всей жизни, рассматриваются новые возможные стратегии
лечения этих инфекций. Кроме того, также требуется параллельный поиск биомаркеров, использование
которых было бы возможно для выбора наиболее приемлемой терапии и установления временных рамок,
когда применение иммунотерапии наиболее эффективно.
Таблица 1 — Сheckpoint-рецепторы
Checkpoint рецептор | Тип эффекторных клеток | Лиганд | Примечание |
TIM3 | Th2-клетки | Galectin 9 на APCs | Галектин 9 индуцирует внутриклеточный поток |
LAG3 | Природные, индуцированные тимусом и индуцированные, | MHC II класса на APCs | LAG3 улучшает функцию клеток Treg. |
CD96 и TIGIT | T-клетки и NK | CD155 на DCs | CD96 и TIGIT вызывают иммунодепрессию, конкурируя с |
BTLA | Т и В-клетки
| HVEM, выраженный большинством | Лигирование TNFSF14 с помощью HVEM может |
TNFSF14 | Интендативные адаптивные иммунные клетки, включая | ||
GITR | Тreg клетки на высоких уровнях, обычные Т-клетки на
| GITRL на APCs | GITR играет ключевую роль, поддерживая клетки CD4 + |
VISTA | Гемопоэтические клетки | Неизвестно | Доклинические исследования с блокадой VISTA |
 LAG3 и PD1
LAG3 и PD1 BTLA
BTLAБелки иммунной контрольной точки при малярии
Малярия — это антропоноз, вызванный простейшими паразитами рода Plasmodium и передающийся
человеку москитами рода Anopheles.
Большинство случаев малярии вызваны Plasmodium falciparum и Plasmodium vivax, а в 2015 году во
всем мире насчитывалось 212 миллионов новых случаев малярии, из которых 429 000 человек были
связаны только с P. Falciparum[11]. За последние 20 лет было разработано и клинически
апробировано более 100 вакцин для борьбы с малярией. Большинство из них были специально
разработаны для таргетного воздействия на паразитов в печеночной стадии или кровяной стадии
развития. Данный метод был основан на индукции защитных антител и CD4 + Т-клеток, хотя несколько
вакцин были разработаны для генерации CD8 + Т-клеточных ответов.
Лучший вариант вакцины, выявленной на сегодняшний день, является вакцина RTS, S / AS01E; однако,
ее эффективность в первый год введения была только 43,6% , а к четвертому году использования
снизилась до 16,8% [12]. Очевидно, что требуется поиск новой тактики лечения, нацеленной на
Очевидно, что требуется поиск новой тактики лечения, нацеленной на
механизмы иммунного воздействия на паразитов. В некоторых исследованиях было показано, что при
малярии на стадии эритроцитарной шизогонии антитела играют ключевую роль в защите организма, о
чем свидетельствует перенос сыворотки от иммунизированных взрослых детям[17]. Было установлено,
что Т-хелперы1 (Th2) имеют ключевое значение для контроля над количеством паразитов в крови и,
таким образом, для предотвращения развития инфекционного процесса [18, 19]. Антитела играют роль
в уничтожении оставшихся паразитов [20]. Антитела и CD8+ Т-клетки принимают участие в
формировании иммунитета, предотвращая повторное заражение [22]. Исследования также показали, что
при малярии активируется апоптоз В-клеток [23] памяти из-за нарушения функций дендритных клеток
(DC) [24]. Это может объяснить, почему применение вакцин не было успешным. И, вероятно, поэтому
Это может объяснить, почему применение вакцин не было успешным. И, вероятно, поэтому
роль экспрессии PD1 в настоящее время расценивается как основной фактор в утрате иммунитета
против малярии.
Малярия и Т-клеточное истощение
В ходе исследований в районах Мали и Кении, эндемичных по малярии, было обнаружено, что у людей,
недавно инфицированных P. falciparum, повышена экспрессия PD1 на CD4+ [26, 27] и CD8+ T-клетках
[27]. Аналогичным образом, повышение процента CD4+ T-клеток у людей с острыми фазами
инфекции, вызванной P. vivax, P. falciparum имело место повышение экспрессии CTLA4, OX40,
связанный с глюкокортикоидом TNFR-родственный белок и CD69 [28], что указывает на роль
регуляторных T-клеток в подавлении иммунитета к малярии и показывает варианты
потенциальных мишеней для управления контрольной точкой. Было показано, что в то время как PDL1,
Было показано, что в то время как PDL1,
экспрессируемый на дендритных клетках, действительно ослабляет иммунные ответы против малярии,
белок PDL2, экспрессируемый на тех же клетках, улучшает иммунные ответы, ингибируя
взаимодействия PDL1-PD1 [9]. Примечательно, что во время острой фазы инфекционного процесса PD1,
как было показано, обусловливал 95%-ную потерю числа и функциональной способности
паразитоспецифических CD8+ Т-клеток, которые необходимы для борьбы с хроническим заболеванием
[21]. Тем самым экспрессия белка PDL2 предохраняет от развития фатальных осложнений малярии
(рис. 2).
Рисунок 2 — PDL2 защищает от летальной малярии и обладает
трансляционным потенциалом (адаптировано Michelle N. Wykes et al. ,
,
2017).
Применение checkpoint-ингибиторов при малярии
Было показано, что мультимерная форма PDL2, связанная с Fc-областью иммуноглобулина (PDL2-Fc)
применима для нивелирования летальной инфекции и увеличивает шанс выживаемости после повторного
инфицирования через несколько месяцев. Кроме того, комбинированная блокада ингибирующих молекул
белка PDL1 и гена активации лимфоцитов 3 (LAG3) с антителами, ускорила выздоровление от острой
нелетальной малярии в стадии эритроцитарной шизогонии, посредством улучшения функции CD4+клеток
и увеличения титров антител [26]. Антитело-опосредованное инициирование передачи сигналов OX40
также увеличивало эффективность CD4+ T-хелперов, клеточного и гуморального иммунитета и, таким
образом, улучшало эффективность уничтожения паразита при нелетальных формах малярийной инфекции
[30].
Таким образом, отдельные белки контрольной точки способствуют развитию патогенеза малярии, и
дальнейшее исследование их применения для терапевтических целей оправдано.
Эти методы лечения могут также иметь применение с целью «оживления» иммунных клеток,
которые, как предполагается, не выполняют свою функцию у людей, проживающих в районах эндемичных
по малярии [26,27]. Это даст возможность формировать стойкий иммунитет к малярии с помощью
вакцин. Кроме того, использование checkpoint-ингибиторов может дополнять применение малярийных
препаратов для выработки долгосрочного иммунитета, как это представлено на примере PDL2-Fc9.
Т-клеточное истощение и белки иммунной контрольной точки при ВИЧ-инфекции
Т-клеточное истощение является отличительной чертой многих хронических вирусных инфекций, включая
ВИЧ. При отсутствии лечения ВИЧ-инфекции наблюдается повышенная экспрессия множественных белков
При отсутствии лечения ВИЧ-инфекции наблюдается повышенная экспрессия множественных белков
иммунной контрольной точки, включая PD1, CTLA4, TIM3 и LAG3, как на CD4 +, так и на CD8 +клетках
[36-38]. После АРТ экспрессия белков иммунной контрольной точки снижается, но остается
повышенной по сравнению с контрольной группой, в которую входят люди, не имеющие ВИЧ-инфекции.
Повышенная экспрессия PD1 преимущественно наблюдается в Т-клетках памяти, тогда как PD1 и CTLA4
экспрессируются регуляторными Т-клетками, а LAG3 экспрессируется в эффекторных Т-клетках памяти
[41] (рис. 3).
Рисунок 3 — Сheckpoints при ВИЧ-инфекции и инфекции вируса
гепатита В (адаптировано Michelle N. Wykes et al., 2017).
Повышенные уровни экспрессии PD1 на общих и ВИЧ-специфических CD8+ Т-клетках при нелеченой
ВИЧ-инфекции впервые были зарегистрированы более 10 лет назад.
В отсутствие AРТ увеличение экспрессии PD1 было связано с ускоренным снижением количества CD4+
Т-клеток в результате острой инфекции и нелеченой хронической инфекции. Многочисленные
наблюдения продемонстрировали четкую связь между экспрессией PD1 на CD4+ или CD8+ Т-клетках и
клиническим исходом.
Таблица 2 — Резюме доклинических или ex vivo исследований в
области инфекционных заболеваний, сообщающих о преимуществах сheckpoint-ингибиторов
Инфекционное заболевание | Тип аффекторных клеток | Ингибиторные белки | Виды мишеней | Результаты |
ВИЧ | CD4+ и CD8+ T клетки | PD1, CTLA4, TIGIT и LAG3 | Люди и мыши | Увеличение ВИЧ-специфических CD8 + Проводятся клинические исследования при злокачественных новообразованиях |
ВГВ | CD4+ и CD8+ T клетки | PD1, CTLA4, 2B4 и TIM3 | Люди (ex vivo), мыши и сурки | PD1, CTLA4 и TIM3, экспрессируемые на CD4 + и CD8 + PD1, CTLA4 и TIM3 улучшают функцию HBV-специфичных CD8 + Т-клеток in vitro |
ВГС | CD4+ и CD8+ T клетки | PD1 и PDL1 | Люди | Экспрессия PD1 повышается в отношении общего Блокада PDL1 восстанавливает функциональную компетентность CTL, специфичных для HCV, |
Туберкулез | CD4+ и CD8+ T клетки | TIM3 | Мыши | Блокада TIM3 восстанавливает функцию Т-клеток и |
Малярия | CD4+ и CD8+ T клетки; B клетки | PD1, PDL1, CTLA4, LAG3 и TIM3 | Мыши | Ускоренное уничтожение паразитов Выживание от летального заболевания Снижение заболеваемости церебральной малярией |
Использование сheckpoint-ингибиторов in vivo для SIV и ВИЧ-инфекции
По результатам исследований, введение антиPD1 антитела SIV-инфицированным макакам-резус привело к
быстрому увеличению вирус-специфических CD8+ Т-клеток с более высоким функциональным качеством и
снижению передачи сигналов интерферона, улучшение проницаемости кишечника [56].
Возможно, что эффективный ответ Т-клеток на анти-PD1-антитело требует наличия антигена, и что,
поскольку АРТ приводит к резкому сокращению вирусных антигенов, функциональный ответ на блокаду
иммунной контрольной точки может быть ограничен в этой ситуации.
Данные исследований свидетельствуют о том, что анти-CTLA4 антитела оказывают существенное влияние
на ВИЧ, которое сохраняется при проведении АРТ. В основе этого лежит другой механизм действия
анти-PD1-антитела, что приводит к снижению РНК ВИЧ в ткани лимфатических узлов. У людей,
инфицированных ВИЧ, LAG3 также высоко экспрессируется на CD4+ и CD8+ Т-клетках в лимфатических
узлах и крови, и это явление напрямую связано с уровнями РНК ВИЧ в плазме, но обратно
пропорционально количеству лимфоцитов CD4+ [59].
Блокада TIGIT и PD1 с анти-TIGIT и anti-PDL1 антителами ex vivo привела к значительному улучшению
ВИЧ-специфической функции CD4+ T-клеток у людей, инфицированных ВИЧ, в том числе и на АРT
[38].
Белки иммунной контрольной точки и устойчивость ВИЧ
В отличие от злокачественных клеток, которые обычно экспрессируют лиганды белков иммунной
контрольной точки, таких как PDL1 [62], у людей, инфицированных ВИЧ, в том числе и при
проведении АРТ, сами белки иммунной контрольной точки избирательно распознают клетки,
инфицированные ВИЧ, которые сохраняются на фоне АРТ [63-64] (рис. 4).
Рисунок 4 — Предполагаемая роль PD1 в формировании и
изменении латентности ВИЧ (адаптировано Michelle N. Wykes et al., 2017).
Это наблюдение имеет большое значение для разработки метода по ликвидации резидуального вируса,
который сохраняется, несмотря на проводимую АРТ, поскольку наличие инфицированных клеткок
является основным препятствием для излечения. Многие исследования показали значительную
Многие исследования показали значительную
корреляцию между частотой встречаемости PD1+ CD4+ T-клеток и PD1+ CD8+ T-клеток с различными
маркерами устойчивости ВИЧ-инфекции к АРТ в крови [63,65,66], лимфатических узлах [67] и
желудочно-кишечном тракте. Белки иммунной контрольной точки, отличные от PD1, также могут
распознавать инфицированные клетки у лиц, получающих АРТ. В недавнем времени было
показано, что ВИЧ может быть значительно обогащен клетками, полученными от людей, инфицированных
ВИЧ и получающих AРT, которые экспрессировали PD1, TIGIT и LAG3 по сравнению с клетками, которые
не экспрессировали ни один из этих белков [64](рис. 3). Внедрение CTLA4, опосредованное вирусным
белком Nef, потенциально может играть роль в сохранении устойчивости ВИЧ в этих клетках [70].
Также было показано, что у человека с диагнозом «метастатическая меланома»,
инфицированного ВИЧ и получающего АРТ, наблюдается значительное увеличение связанной с клетками
ВИЧ-РНК после лечения анти- CTLA4 антителами (Ипилимумаб) [71] и анти-PD1 антителами (Ниволумаб)
[72]. Эти данные еще должны быть подтверждены в клинических исследованиях.
Эти данные еще должны быть подтверждены в клинических исследованиях.
Использование сheckpoint-ингибиторов в качестве стратегии лечения ВИЧ
Изучение доза-зависимой фазы анти-PDL1-терапии было прекращено после введения самой низкой дозы 6
людям с ВИЧ-инфекцией получающим АРТ [73], в связи с развитием токсического воздействия на
сетчатку. На сегодняшний день лишь немногие люди с ВИЧ-инфекцией получили зарегистрированные
препараты сheckpoint-ингибиторов, поскольку лица с ВИЧ-инфекцией были исключены из исследования.
Но в настоящее время использование данных препаратов для ВИЧ-инфицированных людей становится
возможным.
Таким образом, сheckpoint-ингибиторы могут внести существенный вклад в достижение полного
излечения от ВИЧ-инфекции и позволять людям безопасно прекращать АРТ.
Т-клеточное истощение и белки иммунных контрольных точек при HBV-инфекции
При хронической инфекции HBV, не подвергавшейся лечению, общие и HBV-специфичные CD8 + Т-клетки
экспрессируют высокие уровни PD1, CTLA4 и TIM3 [84-86], а при острой инфекции HBV циркулирующие
и внутрипеченочные CD8+ Т-клетки экспрессируют высокие уровни PD1 [87].
Было показано, что наличие лиганда для PD1, PDL1 более высокий на циркулирующих CD14+ моноцитах и
CD19+ B-клетках у лиц с хронической инфекцией HBV, циррозом печени и HCC и, следовательно, может
способствовать постоянному Т-клеточному истощению [88]. Исследования показали, что дефекты
функции Т-клеток при хронической инфекции HBV не ограничиваются увеличением экспрессии белков
иммунной контрольной точки. Внутрипеченочные Т-клетки также проявляют повышенную экспрессию PD1
[87] и TIM3 [84]. Галектин (лиганд TIM3) также был активен на клетках Купфера, что, возможно,
Галектин (лиганд TIM3) также был активен на клетках Купфера, что, возможно,
позволяет поддерживать внутрипеченочное Т-клеточное истощение [84]. Внутрипеченочные CD8+
Т-клетки также экспрессируют другие белки истощения, включая BTLA, и могут продуцировать IL-10,
что дополнительно ингибирует функцию Т-клеток [92].
Применение сheckpoint-ингибиторов в качестве стратегии лечения вирусного гепатита В
Исследования с использованием крови людей, страдающих с хроническим гепатитом В,
продемонстрировали, что ингибирование PD1, CTLA4, 2B4 и TIM3 приводит к увеличению функции
HBV-специфичных CD8+ Т-клеток [83,84,86,93–96]. Применение сheckpoint-ингибиторов в
качестве как монотерапии, так и в сочетании с антивирусными препаратами, может увеличить
продукцию HBV-специфичных CD8+ Т-клеток. А также влиять на выработку антител против HBsAg. Но
А также влиять на выработку антител против HBsAg. Но
при этом существуют значительные риски, в том числе повышенная инфильтрация ткани печени
повторно активированными Т-клеткам, что может вызвать воспаление.
Установлено, что добавление анти-PDL1-антитела к вакцине и энтекавиру по сравнению с одной только
вакциной и энтекавиром привело к значительному увеличению иммунологического и клинического
ответа без развития гепатотоксичности [97]. Недавнее исследование open-label Ниволумаба
(анти-PD1-Ат) с вакциной против HBV и без нее и 20 участниками с подавленной вирусной
хронической инфекцией HBV показало, что Ниволумаб безопасен и хорошо переносится, а один
участник подвергся сероконверсии HBsAg [99].
В настоящее время ведется множество клинических испытаний сheckpoint-ингибиторов у лиц с
хроническим HBV. Таким образом, применение сheckpoint-ингибиторов играет важную роль в лечении
Таким образом, применение сheckpoint-ингибиторов играет важную роль в лечении
вирусного гепатита В, ограничивая повреждение печени при острой инфекции и способствуя
стабилизации процесса при хроническом HBV.
Белки иммунной контрольной точки при туберкулезе
Для формирования невосприимчивости организма к M. tuberculosis необходимы CD4+ Т-клетки. Было
обнаружено, что TBC-специфические CD4+ Т-клетки у лиц с активной туберкулезной инфекцией
продуцируют IFNγ, IL2 и TNF и экспрессируют PD1. Было показано, что функционально
истощенные ТМ3 + Т-клетки накапливаются при наличии хронической туберкулезной инфекции.
Ингибирование TIM3 восстанавливает функции Т-клеток и улучшает контроль за бактериальной
нагрузкой у лиц с хроническим течением туберкулеза. Комбинация препаратов, рутинно используемых
для лечения туберкулеза, с сheckpoint-ингибиторами даст возможность формирования иммунитета
развиваться, при этом, бактериальная нагрузка будет находиться под частичным контролем.
Заключение
Лекарственная терапия малярии, ВИЧ-инфекции, вирусного гепатита В и туберкулеза в настоящее время
является большой проблемой в связи с развитием полирезистентности микроорганизмов, при этом
разработка эффективной вакцины зачастую невозможна. Поэтому применение сheckpoint-ингибиторов
может оказаться абсолютно новой стратегией борьбы с хроническими инфекциями, для которых
по-прежнему отсутствуют эффективные методы лечения, и возможности применения вакцин ограничены.
Однако следует учесть, что применение данных препаратов в свою очередь имеет ограничения и
побочные эффекты. В ряде исследований было выдвинуто предположение, что ингибирование иммунной
контрольной точки также может быть использовано для лечения ряда инфекционных заболеваний, в том
числе в терапии малярии, ВИЧ-инфекции, что несомненно требует подтверждения в клинических
исследованиях.
Источники:
www.nature.com/nri
- Baumeister, S. H., Freeman, G. J., Dranoff, G. & Sharpe, A. H. Coinhibitory pathways in
immunotherapy for cancer. Annu. Rev. Immunol. 34, 539–573 (2016). - Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer
12, 252–264 (2012). - Wherry, E. J. T cell exhaustion. Nat. Immunol. 12, 492–499 (2011).
- Larkin, J. et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N.
Engl. J. Med. 373, 23–34 (2015). - Brahmer, J. R. et al. Phase I study of single-agent anti-programmed death?1 (MDX?1106) in
refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic
correlates. J. Clin. Oncol. 28, 3167–3175 (2010). - Chen, P. L. et al. Analysis of immune signatures in longitudinal tumor samples yields
insight into biomarkers of response and mechanisms of resistance to immune checkpoint
blockade. Cancer Discov. 6, 827–837 (2016). - Kamphorst, A. O. et al. Proliferation of PD?1+ CD8 T cells in peripheral blood after
PD?1?targeted therapy in lung cancer patients. Proc. Natl Acad. Sci. USA 114, 4993–4998
(2017). - Huang, A. C. et al. T?Cell invigoration to tumour burden ratio associated with anti?PD?1
response. Nature 545, 60–65 (2017). - Karunarathne, D. S. et al. Programmed death?1 ligand 2?mediated regulation of the PD?L1 to
PD?1 axis is essential for establishing CD4+ T cell immunity. Immunity 45, 333–345
(2016). - An important study on the mechanism of the protective function of PDL2.
- Cheng, Q., Kyle, D. E. & Gatton, M. L. Artemisinin resistance in Plasmodium falciparum:
a process linked to dormancy? Int. J. Parasitol. Drugs Drug Resist. 2, 249–255 (2012). - World Health Organization. World Malaria Report (WHO, 2016).
- Olotu, A. et al. Four-year efficacy of RTS,S/AS01E and its interaction with malaria
exposure. N. Engl. J. Med. 368, 1111–1120 (2013). - Barragan, A., Kremsner, P. G., Weiss, W., Wahlgren, M. & Carlson, J. Age-related buildup
of humoral immunity against epitopes for rosette formation and agglutination in African
areas of malaria endemicity. Infect. Immun. 66, 4783–4787 (1998). - Bull, P. C. et al. Parasite antigens on the infected red cell surface are targets for
naturally acquired immunity to malaria. Nat. Med. 4, 358–360 (1998). - Soe, S. et al. Premunition against Plasmodium falciparum in a malaria hyperendemic village
in Myanmar. Trans. R. Soc. Trop. Med. Hyg. 95, 81–84 (2001). - Mazier, D. et al. Hepatic phase of malaria is the target of cellular mechanisms induced by
the previous and the subsequent stages. A crucial role for liver nonparenchymal cells.
Immunol. Lett. 25, 65–70 (1990). - Cohen, S., McGregor, I. & Carrington, S. Gamma-globulin and acquired immunity to human
malaria. Nature 192, 733–737 (1961). - Langhorne, J., Simon-Haarhaus, B. & Meding, S. J. The role of CD4+ T cells in the
protective immune response to Plasmodium chabaudi in vivo. Immunol. Lett. 25, 101–107
(1990). - Podoba, J. E. & Stevenson, M. M. CD4+ and CD8+ T lymphocytes both contribute to acquired
immunity to blood-stage Plasmodium chabaudi AS. Infect. Immun. 59, 51–58 (1991). - von der Weid, T., Honarvar, N. & Langhorne, J. Gene-targeted mice lacking B cells are
unable to eliminate a blood stage malaria infection. J. Immunol. 156, 2510–2516
(1996). - Horne-Debets, J. M. et al. PD?1 dependent exhaustion of CD8+ T cells drives chronic malaria.
Cell Rep. 5, 1204–1213 (2013). - The first study to show a role for CD8+ T cells in controlling blood-stage malaria.
- Horne-Debets, J. M. et al. Mice lacking Programmed cell death?1 show a role for CD8+ T cells
in long-term immunity against blood-stage malaria. Sci. Rep. 6, 26210 (2016). - Wykes, M. N., Zhou, Y. H., Liu, X. Q. & Good, M. F. Plasmodium yoelii can ablate
vaccine-induced long-term protection in mice. J. Immunol. 175, 2510–2516 (2005). - Liu, X. Q. et al. Malaria infection alters the expression of B?cell activating factor
resulting in diminished memory antibody responses and survival. Eur. J. Immunol. 42, 3291–3301
(2012). - Pierce, S. K. & Miller, L. H. World Malaria Day 2009: what malaria knows about the
immune system that immunologists still do not. J. Immunol. 182, 5171–5177 (2009). - Butler, N. S. et al. Therapeutic blockade of PD?L1 and LAG?3 rapidly clears established
blood-stage Plasmodium infection. Nat. Immunol. 13, 188–195 (2012). - The first study to show that checkpoint blockade during malaria could improve protective
immunity. - Illingworth, J. et al. Chronic exposure to Plasmodium falciparum is associated with
phenotypic evidence of B and T cell exhaustion. J. Immunol. 190, 1038–1047 (2013). - Goncalves-Lopes, R. M. et al. Surface expression of inhibitory (CTLA?4) and stimulatory
(OX40) receptors by CD4+ regulatory T cell subsets circulating in human malaria. Microbes
Infect. 18, 639–648 (2016). - Hou, N. et al. T?Cell immunoglobulin- and mucin-domain-containing molecule 3 signaling
blockade improves cell-mediated immunity against malaria. J. Infect. Dis. 214, 1547–1556
(2016). - Zander, R. A. et al. PD?1 co?inhibitory and OX40 co?stimulatory crosstalk regulates helper T
cell differentiation and anti-plasmodium humoral immunity. Cell Host Microbe 17, 628–641
(2015). - Hafalla, J. C. et al. The CTLA?4 and PD?1/PD?L1 inhibitory pathways independently regulate
host resistance to plasmodium-induced acute immune pathology. PLoS Pathog. 8, e1002504
(2012). - Hisaeda, H. et al. Resistance of regulatory T cells to glucocorticoid-induced TNFR
family-related protein (GITR) during Plasmodium yoelii infection. Eur. J. Immunol. 35, 3516–3524
(2005). - Lepenies, B. et al. Ligation of B and T lymphocyte attenuator prevents the genesis of
experimental cerebral malaria. J. Immunol. 179, 4093–4100 (2007). - GBD 2015 HIV Collaborators et al. Estimates of global, regional, and national incidence,
prevalence, and mortality of HIV, 1980–2015: the Global Burden of Disease Study 2015.
Lancet HIV 3, e361–e387 (2016). - Deeks, S. G. et al. International AIDS Society global scientific strategy: towards an HIV
cure 2016. Nat. Med. 22, 839–850 (2016). - Trautmann, L. et al. Upregulation of PD?1 expression on HIV-specific CD8+ T cells leads to
reversible immune dysfunction. Nat. Med. 12, 1198–1202 (2006). - One of the first papers to identify PD1 as a marker of T cell exhaustion in HIV infection.
- Kaufmann, D. E. et al. Upregulation of CTLA?4 by HIV-specific CD4+ T cells correlates with
disease progression and defines a reversible immune dysfunction. Nat. Immunol. 8, 1246–1254
(2007). - Chew, G. M. et al. TIGIT marks exhausted T cells, correlates with disease progression, and
serves as a target for immune restoration in HIV and SIV Infection. PLoS Pathog. 12,
e1005349 (2016). - An examination of TIGIT and PD1 in T cell exhaustion in HIV infection both on and off ART.
- Rutishauser, R. et al. Early and delayed antiretroviral therapy (ART) result in comparable
reductions in CD8+ T cell exhaustion marker expression. AIDS Res Hum Retroviruses 33, 658–667
(2017). - Zhang, J. Y. et al. PD?1 up?regulation is correlated with HIV-specific memory CD8+ T?cell
exhaustion in typical progressors but not in long-term nonprogressors. Blood 109, 4671–4678
(2007). - Tian, X. et al. The upregulation of LAG?3 on T cells defines a subpopulation with functional
exhaustion and correlates with disease progression in HIV-infected subjects. J. Immunol.
194, 3873–3882 (2015). - Sauce, D. et al. PD?1 expression on human CD8 T cells depends on both state of
differentiation and activation status. AIDS 21, 2005–2013 (2007). - Day, C. L. et al. PD?1 expression on HIV-specific T cells is associated with T?cell
exhaustion and disease progression. Nature 443, 350–354 (2006). - Petrovas, C. et al. PD?1 is a regulator of virus-specific CD8+ T cell survival in HIV
infection. J. Exp. Med. 203, 2281–2292 (2006). - Leong, Y. A. et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell
follicles. Nat. Immunol. 17, 1187–1196 (2016). - Mylvaganam, G. H. et al. Dynamics of SIV-specific CXCR5+ CD8 T cells during chronic SIV
infection. Proc. Natl Acad. Sci. USA 114, 1976–1981 (2017). - Li, S. et al. Simian immunodeficiency virus-producing cells in follicles are partially
suppressed by CD8+ cells in vivo. J. Virol. 90, 11168–11180 (2016). - Hoffmann, M. et al. Exhaustion of activated CD8 T cells predicts disease progression in
primary HIV?1 Infection. PLoS Pathog. 12, e1005661 (2016). - Shive, C. L. et al. Inflammation perturbs the IL?7 axis, promoting senescence and exhaustion
that broadly characterize immune failure in treated HIV infection. J. Acquir. Immune Def.
Syndr. 71, 483–492 (2016). - Sinha, A. et al. role of T?cell dysfunction, inflammation, and coagulation in microvascular
disease in HIV. J. Am. Heart Assoc. 5, e004243 (2016). - Kelesidis, T. et al. Oxidized lipoproteins are associated with markers of inflammation and
immune activation in HIV?1 infection. AIDS 30, 2625–2633 (2016). - Hurst, J. et al. Immunological biomarkers predict HIV?1 viral rebound after treatment
interruption. Nat. Commun. 6, 8495 (2015). - The first publication to show a functional link between expression of immune checkpoint
proteins on T cells prior to ART and the time to rebound after cessation of ART. An
important observation in determining the role of immune checkpoint proteins and HIV cure or
remission. - Akhmetzyanova, I. et al. PD?L1 expression on retrovirus-infected cells mediates immune
escape from CD8+ T cell killing. PLoS Pathog. 11, e1005224 (2015). - Velu, V. et al. Enhancing SIV-specific immunity in vivo by PD?1 blockade. Nature 458, 206–210
(2009). - Dyavar Shetty, R. et al. PD?1 blockade during chronic SIV infection reduces hyperimmune
activation and microbial translocation in rhesus macaques. J. Clin. Invest. 122, 1712–1716
(2012). - Mylvaganam, G. H. et al. PD?1 blockade synergizes with ART for restoring anti-viral CD8 T
cell function and possibly destabilizing the viral reservoir in SIV infected macaques
[Abstract 9016]. Presented at the International AIDS Conference, Durban, South Africa, July
2016. - Gill, A. L. et al. Programed death?1/programed death-ligand 1 expression in lymph nodes of
HIV infected patients: results of a pilot safety study in rhesus macaques using
anti-programed death-ligand 1 (Avelumab). AIDS 30, 2487–2493 (2016). - Cecchinato, V. et al. Immune activation driven by CTLA?4 blockade augments viral replication
at mucosal sites in simian immunodeficiency virus infection. J. Immunol. 180, 5439–5447
(2008). - Hryniewicz, A. et al. CTLA?4 blockade decreases TGF- β, IDO, and viral RNA expression
in tissues of SIVmac251?infected macaques. Blood 108, 3834–3842 (2006). - Tauriainen, J. et al. Perturbed CD8+ T cell TIGIT/ CD226/PVR axis despite early initiation
of antiretroviral treatment in HIV infected individuals. Sci. Rep. 7, 40354 (2017). - Anderson, A. C., Joller, N. & Kuchroo, V. K. Lag?3, Tim?3, and TIGIT: co?inhibitory
receptors with specialized functions in immune regulation. Immunity 44, 989–1004
(2016). - Chatterjee, S. et al. A humanized antibody for imaging immune checkpoint ligand PD?L1
expression in tumors. Oncotarget 7, 10215–10227 (2016). - Chomont, N. et al. HIV reservoir size and persistence are driven by T cell survival and
homeostatic proliferation. Nat. Med. 15, 893–900 (2009). The first demonstration of
the relationship between PD1 and HIV persistence in patients on ART. - Fromentin, R. et al. CD4+ T cells expressing PD?1, TIGIT and LAG?3 contribute to HIV
persistence during ART. PLoS Pathog. 12, e1005761 (2016). An important paper demonstrating
that multiple immune checkpoint markers, not just PD1, are involved in HIV persistence in
patients on ART. - Hatano, H. et al. Cell-based measures of viral persistence are associated with immune
activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells.. J. Infect.
Dis. 208, 50–56 (2013). - Cockerham, L. R. et al. Programmed death?1 expression on CD4+ and CD8+ T cells in treated
and untreated HIV disease. AIDS 28, 1749–1758 (2014). - Banga, R. et al. PD?1+ and follicular helper T cells are responsible for persistent HIV?1
transcription in treated aviremic individuals. Nat. Med. 22, 754–761 (2016). - The first demonstration that TFH cells that express high levels of PD1 are an important
reservoir for HIV in patients on ART. - Khoury, G. et al. HIV persistence and T?cell activation in blood, rectal and lymph node
tissue in HIV-infected individuals receiving suppressive ART. J. Infect. Dis. 215, 911–919
(2017). - El?Far, M. et al. Nef promotes evasion of human immunodeficiency virus type 1?infected cells
from the CTLA?4?mediated inhibition of T?cell activation. J. Gen. Virol. 96, 1463–1477
(2015). - El?Far, M. et al. Down-regulation of CTLA?4 by HIV?1 Nef protein. PLoS ONE 8, e54295
(2013). - Wightman, F. et al. Effect of ipilimumab on the HIV reservoir in an HIV-infected individual
with metastatic melanoma. AIDS 29, 504–506 (2015). - Van der Sluis, R. M. et al. Anti?PD?1 disrupts HIV latency in non-proliferating but not in
proliferating T?cells [Abstract OA3?3]. J. Virus Eradication 3, Suppl. 1 (2017). - Gay, C. L. et al. Clinical trial of the anti?PD?L1 antibody BMS?936559 in HIV?1 infected
participants on suppressive antiretroviral therapy. J. Infect. Dis. 215, 1725–1733
(2017). - Rasmussen, T. A., Anderson, J. L., Wightman, F. & Lewin, S. R. Cancer therapies in HIV
cure research. Curr. Opin. HIV AIDS 12, 96–104 (2017). - Romano, E. et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex
vivo by nonclassical monocytes in melanoma patients. Proc. Natl Acad. Sci. USA 112, 6140–6145
(2015). - Dahan, R. et al. FcγRs modulate the anti-tumor activity of antibodies targeting the
PD?1/PD?L1 axis. Cancer Cell 28, 285–295 (2015). - An interesting paper exploring the future direction of immune checkpoint blockers with
modifications to the Fc tail so that the antibody activates Fc receptors. - Zhou, J. et al. PD1?based DNA vaccine amplifies HIV?1 GAG-specific CD8+ T cells in mice. J.
Clin. Invest. 123, 2629–2642 (2013). - Schweitzer, A., Horn, J., Mikolajczyk, R. T., Krause, G. & Ott, J. J. Estimations of
worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data
published between 1965 and 2013. Lancet 386, 1546–1555 (2015). - Stanaway, J. D. et al. The global burden of viral hepatitis from 1990 to 2013: findings from
the Global Burden of Disease Study 2013. Lancet 388, 1081–1088 (2016). - Revill, P., Testoni, B., Locarnini, S. & Zoulim, F. Global strategies are required to
cure and eliminate HBV infection. Nat. Rev. Gastroenterol. Hepatol. 13, 239–248
(2016). - Guidotti, L. G., Isogawa, M. & Chisari, F. V. Host-virus interactions in hepatitis B
virus infection. Curr. Opin. Immunol. 36, 61–66 (2015). - Chang, J. J. et al. The phenotype of hepatitis B virus-specific T cells differ in the liver
and blood in chronic hepatitis B virus infection. Hepatology 46, 1332–1340 (2007). - Boni, C. et al. Characterization of hepatitis B virus (HBV)-specific T?cell dysfunction in
chronic HBV infection. J. Virol. 81, 4215–4225 (2007). - Nebbia, G. et al. Upregulation of the Tim?3/galectin?9 pathway of T cell exhaustion in
chronic hepatitis B virus infection. PLoS ONE 7, e47648 (2012). - Bengsch, B., Martin, B. & Thimme, R. Restoration of HBV-specific CD8+ T cell function by
PD?1 blockade in inactive carrier patients is linked to T cell differentiation. J. Hepatol.
61, 1212–1219 (2014). - Schurich, A. et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen?4 on
apoptosis-prone CD8 T cells in persistent hepatitis B virus infection. Hepatology 53, 1494–1503
(2011). - Zhang, Z. et al. Dynamic programmed death 1 expression by virus-specific CD8 T cells
correlates with the outcome of acute hepatitis B. Gastroenterology 134, 1938–1949
(2008). - A demonstration of the importance of PD1 expression in acute HBV infection.
- Huang, Z. Y. et al. Clinical significance of dynamics of programmed death ligand?1
expression on circulating CD14+ monocytes and CD19+ B Cells with the progression of
Hepatitis B virus infection. Viral Immunol. 30, 224–231 (2017). - Raziorrouh, B. et al. Inhibitory phenotype of HBV-specific CD4+ T?cells is characterized by
high PD?1 expression but absent coregulation of multiple inhibitory molecules. PLoS ONE 9,
e105703 (2014). - Fisicaro, P. et al. Targeting mitochondrial dysfunction can restore antiviral activity of
exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat. Med. 23, 327–336
(2017). - Maini, M. K. et al. The role of virus-specific CD8+ cells in liver damage and viral control
during persistent hepatitis B virus infection. J. Exp. Med. 191, 1269–1280 (2000). - Wang, H. et al. Hepatic expansion of virus-specific CD8+BTLA+ T cells with regulatory
properties in chronic hepatitis B virus infection. Cell. Immunol. 311, 36–45 (2017). - Fisicaro, P. et al. Antiviral intrahepatic T?cell responses can be restored by blocking
programmed death?1 pathway in chronic hepatitis B. Gastroenterology 138, 682–693
(2010). - Raziorrouh, B. et al. The immunoregulatory role of CD244 in chronic hepatitis B infection
and its inhibitory potential on virus-specific CD8+ T?cell function. Hepatology 52, 1934–1947
(2010). - Isogawa, M., Furuichi, Y. & Chisari, F. V. Oscillating CD8+ T cell effector functions
after antigen recognition in the liver. Immunity 23, 53–63 (2005). - Maier, H., Isogawa, M., Freeman, G. J. & Chisari, F. V. PD?1:PD?L1 interactions
contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver.
J. Immunol. 178, 2714–2720 (2007). - Zhang, E. et al. The expression of PD?1 ligands and their involvement in regulation of T
cell functions in acute and chronic woodchuck hepatitis virus infection. PLoS ONE 6, e26196
(2011). - Liu, J. et al. Enhancing virus-specific immunity in vivo by combining therapeutic
vaccination and PD?L1 blockade in chronic hepadnaviral infection. PLoS Pathog. 10, e1003856
(2014). - A study showing the effect of anti?PDL1 antibody in an animal model of HBV; it reduced viral
rebound after cessation of antiviral therapy and increased antibodies to HBV surface
antigen. - Gane, E. J. et al. A phase 1 study evaluating anti?PD?1 treatment with or without GS?4774 in
HBeAg negative chronic hepatitis B patients [Abstract]. Eur. Associ. Study Liver Dis.
(2017). - El-Khoueiry, A. B. et al. Nivolumab in patients with advanced hepatocellular carcinoma
(CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion
trial. Lancet http://dx.doi.org/10.1016/ S0140-6736(17)31046-2 (2017). - Pollock, K. M. et al. PD?1 expression and cytokine secretion profiles of Mycobacterium
tuberculosis-specific CD4+ T?cell subsets; potential correlates of containment in HIV?TB
co?Infection. PLoS ONE 11, e0146905 (2016). - Boer, M. C. et al. KLRG1 and PD?1 expression are increased on T?cells following
tuberculosis-treatment and identify cells with different proliferative capacities in
BCG-vaccinated adults. Tuberculosis (Edinb). 97, 163–171 (2016) - Barber, D. L., Mayer-Barber, K. D., Feng, C. G., Sharpe, A. H. & Sher, A. CD4 T cells
promote rather than control tuberculosis in the absence of PD?1?mediated inhibition. J.
Immunol. 186, 1598–1607 (2011). - Tousif, S. et al. T cells from programmed death?1 deficient mice respond poorly to
Mycobacterium tuberculosis infection. PLoS ONE 6, e19864 (2011). - Lazar-Molnar, E. et al. Programmed death?1 (PD?1)- deficient mice are extraordinarily
sensitive to tuberculosis. Proc. Natl Acad. Sci. USA 107, 13402–13407 (2010). - Jayaraman, P. et al. TIM3 mediates T cell exhaustion during Mycobacterium tuberculosis
infection. PLoS Pathog. 12, e1005490 (2016). - Sotgiu, G., Centis, R., D’Ambrosio, L. & Migliori, G. B. Tuberculosis treatment
and drug regimens. Cold Spring Harb. Perspect Med. 5, a017822 (2015). - Attanasio, J. & Wherry, E. J. Costimulatory and coinhibitory receptor pathways in
infectious disease. Immunity 44, 1052–1068 (2016). - Harding, F. A., McArthur, J. G., Gross, J. A., Raulet, D. H. & Allison, J. P.
CD28?mediated signalling co?stimulates murine T cells and prevents induction of anergy in
T?cell clones. Nature 356, 607–609 (1992). - Krummel, M. F. & Allison, J. P. CD28 and CTLA?4 have opposing effects on the response of
T cells to stimulation. J. Exp. Med. 182, 459–465 (1995). - A seminal study of CTLA4.
- Freeman, G. J. et al. Engagement of the PD?1 immunoinhibitory receptor by a novel B7 family
member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034
(2000). - A seminal study of the PD1–PDL1 interaction.
- Keir, M. E., Butte, M. J., Freeman, G. J. & Sharpe, A. H. PD?1 and its ligands in
tolerance and immunity. Annu. Rev. Immunol. 26, 677–704 (2008). - Chemnitz, J. M., Parry, R. V., Nichols, K. E., June, C. H. & Riley, J. L. SHP?1 and
SHP?2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon
primary human T cell stimulation, but onlyreceptor ligation prevents T cell activation. J.
Immunol. 173, 945–954 (2004). - Francisco, L. M. et al. PD?L1 regulates the development, maintenance, and function of
induced regulatory T cells. J. Exp. Med. 206, 3015–3029 (2009). - Kamphorst, A. O. et al. Rescue of exhausted CD8 T cells by PD?1?targeted therapies is
CD28?dependent. Science 355, 1423–1427 (2017). - An important study highlighting the balance between immune activation and immune
suppression. - Yamazaki, T. et al. Expression of programmed death 1 ligands by murine T cells and APC. J.
Immunol. 169, 5538–5545 (2002). - Keir, M. E. et al. Tissue expression of PD?L1 mediates peripheral T cell tolerance. J. Exp.
Med. 203, 883–895 (2006). - Brown, J. A. et al. Blockade of programmed death?1 ligands on dendritic cells enhances T
cell activation and cytokine production. J. Immunol. 170, 1257–1266 (2003). - Liang, S. C. et al. Regulation of PD?1, PD?L1, and PD?L2 expression during normal and
autoimmune responses. Eur. J. Immunol. 33, 2706–2716 (2003). - Balar, A. V. et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients
with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase
2 trial. Lancet 389, 67–76 (2017). - Rittmeyer, A. et al. Atezolizumab versus docetaxel in patients with previously treated
non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled
trial. Lancet 389, 255–265 (2017). - Homet Moreno, B. , Mok, S., Comin-Anduix, B., Hu?Lieskovan, S. & Ribas, A. Combined
treatment with dabrafenib and trametinib with immune-stimulating antibodies for BRAF mutant
melanoma. Oncoimmunol. 5, e1052212 (2016). - Latchman, Y. et al. PD?L2 is a second ligand for PD?1 and inhibits T cell activation. Nat.
Immunol. 2, 261–268 (2001). - Zhang, Y. et al. Regulation of T cell activation and tolerance by PDL2. Proc. Natl Acad.
Sci. USA 103, 11695–11700 (2006). - Tseng, S. Y. et al. B7?DC, a new dendritic cell molecule with potent costimulatory
properties for T cells. J. Exp. Med. 193, 839–846 (2001). - First study to highlight a protective role for PDL2.
- Shin, T. et al. Cooperative B7?1/2 (CD80/CD86) and B7?DC costimulation of CD4+ T cells
independent of the PD?1 receptor. J. Exp. Med. 198, 31–38 (2003). - Shin, T. et al. In vivo costimulatory role of B7?DC in tuning T. helper cell 1 and cytotoxic
T. lymphocyte responses. J. Exp. Med. 201, 1531–1541 (2005). - Michot, J. M. et al. Immune-related adverse events with immune checkpoint blockade: a
comprehensive review. Eur. J. Cancer 54, 139–148 (2016). - Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma.
N. Engl. J. Med. 363, 711–723 (2010). - Topalian, S. L. et al. Safety, activity, and immune correlates of anti?PD?1 antibody in
cancer. N. Engl. J. Med. 366, 2443–2454 (2012). - Brahmer, J. R. et al. Safety and activity of anti?PD?L1 antibody in patients with advanced
cancer. N. Engl. J. Med. 366, 2455–2465 (2012). - Weber, J. S., Postow, M., Lao, C. D. & Schadendorf, D. Management of adverse events
following treatment with anti-programmed death?1 agents. Oncologist 21, 1230–1240
(2016). - Topalian, S. L. et al. Survival, durable tumor remission, and long-term safety in patients
with advanced melanoma receiving nivolumab. J. Clin. Oncol. 32, 1020–1030 (2014). - Barber, D. L. et al. Restoring function in exhausted CD8 T cells during chronic viral
infection. Nature 439, 682–687 (2006). - Wherry, E. J. et al. Molecular signature of CD8+ T cell exhaustion during chronic viral
infection. Immunity 27, 670–684 (2007). - Youngblood, B. et al. Cutting edge: prolonged exposure to HIV reinforces a poised epigenetic
program for PD?1 expression in virus-specific CD8 T cells. J. Immunol. 191, 540–544
(2013). - Ahn, E. et al. Demethylation of the PD?1 promoter is imprinted during the effector phase of
CD8 T cell exhaustion. J. Virol. 90, 8934–8946 (2016). - Pauken, K. E. et al. Epigenetic stability of exhausted T cells limits durability of
reinvigoration by PD?1 blockade. Science 354, 1160–1165 (2016). - Sen, D. R. et al. The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169
(2016). - An important paper defining how T cell exhaustion is controlled at the epigenetic level.
- Zhu, C. et al. The Tim?3 ligand galectin?9 negatively regulates T helper type 1 immunity.
Nat. Immunol. 6, 1245–1252 (2005). - Zhou, Q. et al. Coexpression of Tim?3 and PD?1 identifies a CD8+ T?cell exhaustion phenotype
in mice with disseminated acute myelogenous leukemia. Blood 117, 4501–4510 (2011). - Sakuishi, K. et al. Targeting Tim?3 and PD?1 pathways to reverse T cell exhaustion and
restore anti-tumor immunity. J. Exp. Med. 207, 2187–2194 (2010). - Huang, C. T. et al. Role of LAG?3 in regulatory T cells. Immunity 21, 503–513
(2004). - Woo, S. R. et al. Immune inhibitory molecules LAG?3 and PD?1 synergistically regulate T?cell
function to promote tumoral immune escape. Cancer Res. 72, 917–927 (2012). - Dougall, W. C., Kurtulus, S., Smyth, M. J. & Anderson, A. C. TIGIT and CD96: new
checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 276, 112–120
(2017). - Lozano, E., Dominguez-Villar, M., Kuchroo, V. & Hafler, D. A. The TIGIT/CD226 axis
regulates human T cell function. J. Immunol. 188, 3869–3875 (2012). - Yu, X. et al. The surface protein TIGIT suppresses T cell activation by promoting the
generation of mature immunoregulatory dendritic cells. Nat. Immunol. 10, 48–57 (2009). - Blake, S. J., Dougall, W. C., Miles, J. J., Teng, M. W. & Smyth, M. J. Molecular
pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin. Cancer Res. 22, 5183–5188
(2016). - Gonzalez, L. C. et al. A coreceptor interaction between the CD28 and TNF receptor family
members B and T lymphocyte attenuator and herpesvirus entry mediator. Proc. Natl Acad. Sci.
USA 102, 1116–1121 (2005). - Sedy, J. R. et al. B and T lymphocyte attenuator regulates T cell activation through
interaction with herpesvirus entry mediator. Nat. Immunol. 6, 90–98 (2005). - Cai, G. & Freeman, G. J. The CD160, BTLA, LIGHT/ HVEM pathway: a bidirectional switch
regulating T?cell activation. Immunol. Rev. 229, 244–258 (2009). - Fourcade, J. et al. CD8+ T cells specific for tumor antigens can be rendered dysfunctional
by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and
PD?1. Cancer Res. 72, 887–896 (2012). - Ephrem, A. et al. Modulation of Treg cells/T effector function by GITR signaling is
context-dependent. Eur. J. Immunol. 43, 2421–2429 (2013). - Shimizu, J., Yamazaki, S., Takahashi, T., Ishida, Y. & Sakaguchi, S. Stimulation of
CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol.
3, 135–142 (2002). - Schaer, D. A., Cohen, A. D. & Wolchok, J. D. Anti-GITR antibodies — potential
clinical applications for tumor immunotherapy. Curr. Opin. Investig. Drugs 11, 1378–1386
(2010). - Mitsui, J. et al. Two distinct mechanisms of augmented antitumor activity by modulation of
immunostimulatory/inhibitory signals. Clin. Cancer Res. 16, 2781–2791 (2010). - Deng, J., Le Mercier, I., Kuta, A. & Noelle, R. J. A new VISTA on combination therapy
for negative checkpoint regulator blockade. J. Immunother. Cancer 4, 86 (2016). - Gao, J. et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab
therapy in patients with prostate cancer. Nat. Med, 23, 551–555 (2017). - Freeman, G. J., Wherry, E. J., Ahmed, R. & Sharpe, A. H. Reinvigorating exhausted
HIV-specific T cells via PD?1?PD?1 ligand blockade. J. Exp. Med. 203, 2223–2227
(2006). - Kaufmann, D. E. & Walker, B. D. PD?1 and CTLA?4 inhibitory cosignaling pathways in HIV
infection and the potential for therapeutic intervention. J. Immunol. 182, 5891–5897
(2009). - Golden-Mason, L. et al. Upregulation of PD?1 expression on circulating and intrahepatic
hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J.
Virol. 81, 9249–9258 (2007). - Nakamoto, N. et al. Functional restoration of HCV-specific CD8 T cells by PD?1 blockade is
defined by PD?1 expression and compartmentalization. Gastroenterology 134, 1927–1937
(2008). - Lukens, J. R., Cruise, M. W., Lassen, M. G. & Hahn, Y. S. Blockade of PD?1/B7?h2
interaction restores effector CD8+ T cell responses in a hepatitis C virus core murine
model. J. Immunol. 180, 4875–4884 (2008) - Urbani, S. et al. Restoration of HCV-specific T cell functions by PD?1/PD?L1 blockade in HCV
infection: effect of viremia levels and antiviral treatment. J. Hepatol. 48, 548–558
(2008). - Rutigliano, J. A. et al. Highly pathological influenza A virus infection is associated with
augmented expression of PD?1 by functionally compromised virus-specific CD8+ T cells. J.
Virol. 88, 1636–1651 (2014). - Sharma, S. et al. T cell immunoglobulin and mucin protein?3 (Tim?3)/Galectin?9 interaction
regulates influenza A virus-specific humoral and CD8 T?cell responses. Proc. Natl Acad. Sci.
USA 108, 19001–19006 (2011). - Xu, D. et al. A potential new pathway for PD?L1 costimulation of the CD8?T cell response to
Listeria monocytogenes infection. PLoS ONE 8, e56539 (2013). - Bhadra, R., Gigley, J. P., Weiss, L. M. & Khan, I. A. Control of Toxoplasma reactivation
by rescue of dysfunctional CD8+ T?cell response via PD?1?PDL?1 blockade. Proc. Natl Acad.
Sci. USA 108, 9196–9201 (2011). - Mou, Z. et al. Parasite-derived arginase influences secondary anti-Leishmania immunity by
regulating programmed cell death?1?mediated CD4+ T cell exhaustion. J. Immunol. 190, 3380–3389
(2013). - Esch, K. J., Juelsgaard, R., Martinez, P. A., Jones, D. E. & Petersen, C. A. Programmed
death 1?mediated T cell exhaustion during visceral leishmaniasis impairs phagocyte function.
J. Immunol. 191, 5542–5550 (2013). - Joshi, T., Rodriguez, S., Perovic, V., Cockburn, I. A. & Stager, S. B7?h2 blockade
increases survival of dysfunctional CD8+ T cells and confers protection against Leishmania
donovani infections. PLoS Pathog. 5, e1000431 (2009).
 J. Clin. Oncol. 28, 3167–3175 (2010).
J. Clin. Oncol. 28, 3167–3175 (2010). Immunity 45, 333–345
Immunity 45, 333–345 Infect. Immun. 66, 4783–4787 (1998).
Infect. Immun. 66, 4783–4787 (1998). Nature 192, 733–737 (1961).
Nature 192, 733–737 (1961).
 K. & Miller, L. H. World Malaria Day 2009: what malaria knows about the
K. & Miller, L. H. World Malaria Day 2009: what malaria knows about the Microbes
Microbes et al. Resistance of regulatory T cells to glucocorticoid-induced TNFR
et al. Resistance of regulatory T cells to glucocorticoid-induced TNFR

 J. Immunol.
J. Immunol. H. et al. Dynamics of SIV-specific CXCR5+ CD8 T cells during chronic SIV
H. et al. Dynamics of SIV-specific CXCR5+ CD8 T cells during chronic SIV et al. role of T?cell dysfunction, inflammation, and coagulation in microvascular
et al. role of T?cell dysfunction, inflammation, and coagulation in microvascular
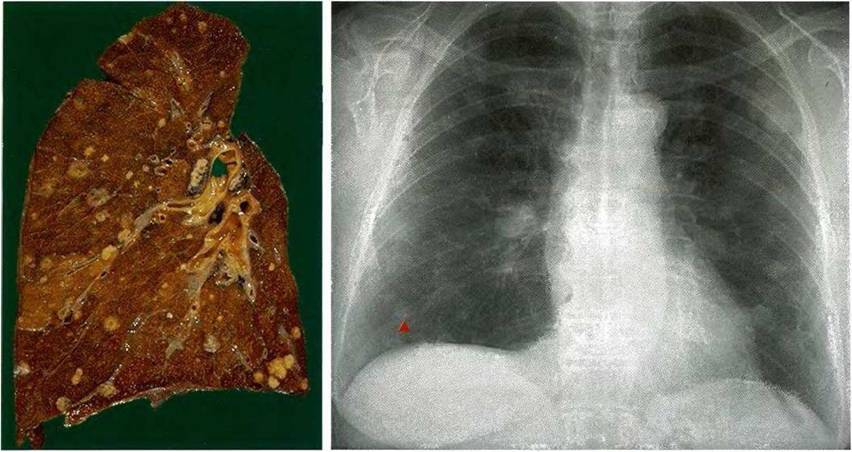 Presented at the International AIDS Conference, Durban, South Africa, July
Presented at the International AIDS Conference, Durban, South Africa, July
 The first demonstration of
The first demonstration of AIDS 28, 1749–1758 (2014).
AIDS 28, 1749–1758 (2014). J. Gen. Virol. 96, 1463–1477
J. Gen. Virol. 96, 1463–1477

 Nat. Rev. Gastroenterol. Hepatol. 13, 239–248
Nat. Rev. Gastroenterol. Hepatol. 13, 239–248 PLoS ONE 7, e47648 (2012).
PLoS ONE 7, e47648 (2012).
 The role of virus-specific CD8+ cells in liver damage and viral control
The role of virus-specific CD8+ cells in liver damage and viral control
 PLoS Pathog. 10, e1003856
PLoS Pathog. 10, e1003856

 & Wherry, E. J. Costimulatory and coinhibitory receptor pathways in
& Wherry, E. J. Costimulatory and coinhibitory receptor pathways in J. Exp. Med. 192, 1027–1034
J. Exp. Med. 192, 1027–1034 J. Exp. Med. 206, 3015–3029 (2009).
J. Exp. Med. 206, 3015–3029 (2009). J. Immunol. 170, 1257–1266 (2003).
J. Immunol. 170, 1257–1266 (2003).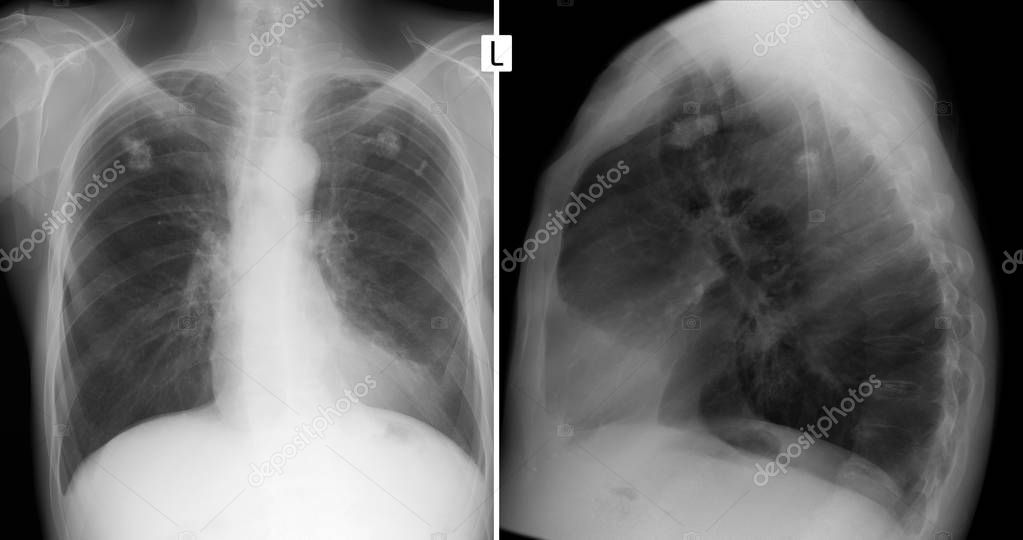 , Mok, S., Comin-Anduix, B., Hu?Lieskovan, S. & Ribas, A. Combined
, Mok, S., Comin-Anduix, B., Hu?Lieskovan, S. & Ribas, A. Combined
 N. Engl. J. Med. 366, 2443–2454 (2012).
N. Engl. J. Med. 366, 2443–2454 (2012).
 R. et al. The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169
R. et al. The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169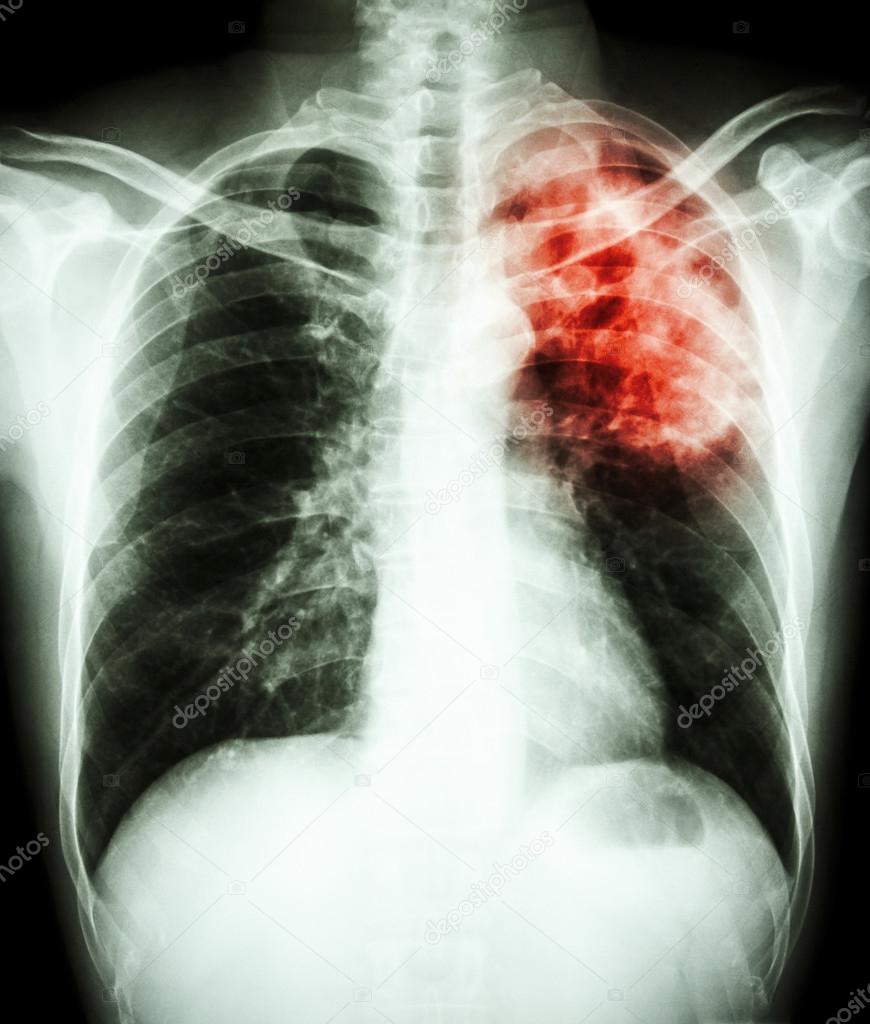 T. et al. Role of LAG?3 in regulatory T cells. Immunity 21, 503–513
T. et al. Role of LAG?3 in regulatory T cells. Immunity 21, 503–513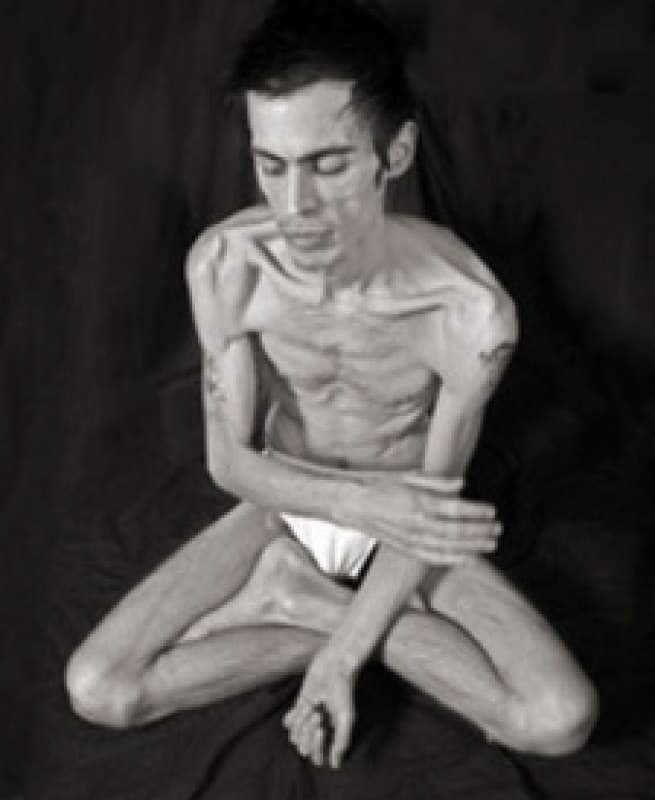 Nat. Immunol. 10, 48–57 (2009).
Nat. Immunol. 10, 48–57 (2009). Immunol. Rev. 229, 244–258 (2009).
Immunol. Rev. 229, 244–258 (2009). D. Anti-GITR antibodies — potential
D. Anti-GITR antibodies — potential J., Wherry, E. J., Ahmed, R. & Sharpe, A. H. Reinvigorating exhausted
J., Wherry, E. J., Ahmed, R. & Sharpe, A. H. Reinvigorating exhausted Gastroenterology 134, 1927–1937
Gastroenterology 134, 1927–1937
 J. Immunol. 190, 3380–3389
J. Immunol. 190, 3380–3389
Материал подготовлен НОИМТОиР: клинический ординатор Бриш
Н.А.
Бактерия ТБ может подавлять иммунную систему и распространять болезнь
Ученые хорошо понимают, как бактерия, вызывающая туберкулез, одну из основных причин смерти в мире, уклоняется от иммунной системы, чтобы закрепиться в организме.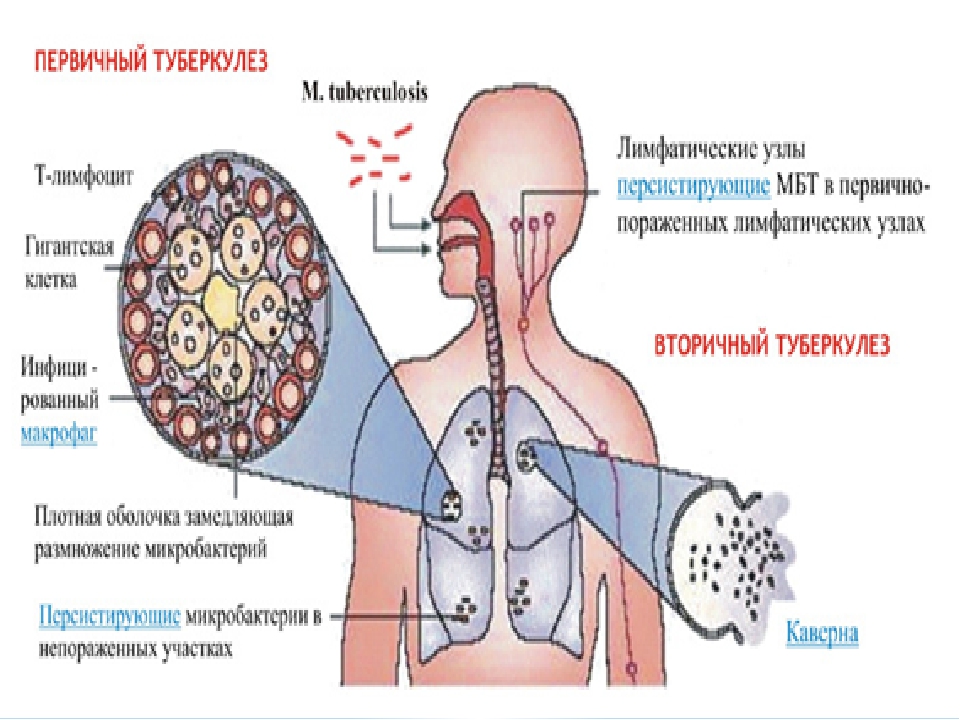 Однако до сих пор не выяснена роль патогена в подрыве иммунной системы, способствующей распространению болезни. Теперь новое исследование обнаруживает доказательства того, что эта болезнетворная бактерия нарушает регуляторные пути в лейкоцитах, которые ограничивают разрушение тканей.
Однако до сих пор не выяснена роль патогена в подрыве иммунной системы, способствующей распространению болезни. Теперь новое исследование обнаруживает доказательства того, что эта болезнетворная бактерия нарушает регуляторные пути в лейкоцитах, которые ограничивают разрушение тканей.
Поделиться на Pinterest В новом исследовании ученые показали, что бактерия, вызывающая туберкулез, может подавлять иммунную систему и распространять болезнь.
Исследование, проведенное учеными из Саутгемптонского университета в Великобритании, опубликовано в журнале PLOS Pathogens .
Туберкулез (ТБ) — инфекционное заболевание, вызываемое микроорганизмом Mycobacterium tuberculosis . Заболевание входит в десятку основных причин смерти во всем мире и в основном поражает легкие.
По данным Всемирной организации здравоохранения (ВОЗ), туберкулез стал причиной заболевания 10,4 миллиона человек и стал причиной 1,8 миллиона смертей в 2015 году.
Основной путь заражения туберкулезом — вдыхание воздуха, содержащего туберкулез. бактерия, которая попадает туда, когда соседний инфицированный человек кашляет, сплевывает или чихает.
бактерия, которая попадает туда, когда соседний инфицированный человек кашляет, сплевывает или чихает.
ТБ можно вылечить и предотвратить, и, по оценке ВОЗ, с 2000 по 2015 год было спасено 49 миллионов жизней благодаря эффективной диагностике и лечению этой болезни.
В своей исследовательской работе исследователи объясняют, что одна из особенностей, делающих туберкулез очень заразным, заключается в том, что он разрушает легочную ткань и создает полости в легких.
Известно, что полости в легких, возникающие при туберкулезе, являются результатом активности ферментов, разрушающих ткани, в частности фермента, называемого матриксной металлопротеиназой-1 (ММП-1), который выделяется из иммунных клеток, называемых макрофагами.
Макрофаги — это белые кровяные тельца, которые нацелены, поглощают и уничтожают нежелательные агенты, такие как патогены и нездоровые клетки человека.
Организм нуждается в ММП-1, чтобы помочь ремоделировать легочную ткань (например, при заживлении ран), но если фермента будет слишком много, это может привести к разрушению ткани.
Команда отмечает, что хотя сигнальные пути, которые контролируют MMP-1 в макрофагах, уже идентифицированы, мало что известно о том, что происходит с такими путями в тканях, инфицированных туберкулезом.
Таким образом, исследователи намеревались узнать больше, изучая молекулярные эффекты бактерии ТБ на изолированные макрофаги.
Они обнаружили, что бактерия TB подавляет сигнальный путь, называемый PI3K / AKT / mTORC1, чтобы заставить макрофаги высвобождать больше MMP-1. Этот путь помогает регулировать MMP-1 так, чтобы высвобождалось ровно столько, сколько нужно для ремоделирования ткани, не вызывая разрушения ткани.
Чтобы подтвердить, что подавление этого пути происходит у людей с туберкулезом, команда исследовала макрофаги в легочной ткани, взятой у инфицированных пациентов.
Они обнаружили, что ген, который необходимо включить для ограничения продукции MMP-1, не проявляет активности у больных туберкулезом, что позволяет предположить, что это могло быть причиной разрушения легочной ткани у этих пациентов.
Изучая изолированные макрофаги, исследователи также обнаружили, что бактерия TB нарушает другой сигнальный путь, называемый киназой, взаимодействующей с MAP-киназой (MNK), в результате чего также увеличивается продукция MMP-1.
Авторы отмечают, что их открытие является первым доказательством связи между путем MNK и продукцией фермента MMP-1.
Важным следствием исследования, которое подчеркивают авторы, является возможность того, что новые и существующие противотуберкулезные препараты непреднамеренно подавляют идентифицированные ими пути.В таком случае такие лекарства могут действительно способствовать распространению туберкулеза.
«Туберкулез эволюционировал вместе с людьми, чтобы стать основным патогеном, и мы определяем механизм, с помощью которого бактерии отключают« тормоза »иммунной системы, вызывая разрушение и распространение легких».
Первый автор Д-р Пейшенс Т. Брейс, Саутгемптонский университет
Узнайте, как соединение, обнаруженное в почвенных бактериях, может давать новые противотуберкулезные препараты.
Истощение альвеолярных макрофагов оказывает защитное действие при туберкулезе легких у мышей
Реферат
Mycobacterium tuberculosis Бациллы — это внутриклеточные организмы, которые обитают в фагосомах альвеолярных макрофагов (AM).Чтобы определить in vivo роль истощения AM в защите хозяина от инфекции M. tuberculosis , мышей с туберкулезом легких, вызванным интраназальным введением вирулентного M. tuberculosis , вводили интраназально любым дихлорметилендифосфонатом, инкапсулированным в липосомы (AM —). мыши), липосомы или физиологический раствор (мыши AM + ). Мыши AM — были полностью защищены от летальности, которая была связана с уменьшением роста микобактерий в легких и печени и поляризованной продукцией цитокинов типа 1 в легочной ткани и спленоцитами, стимулируемыми ex vivo.Мыши AM — демонстрировали недостаточное образование гранулем, но были более способны привлекать и активировать Т-клетки в легкие и имели повышенное количество легочных полиморфно-ядерных клеток. Эти данные демонстрируют, что истощение AM оказывает защитное действие при туберкулезе легких.
Эти данные демонстрируют, что истощение AM оказывает защитное действие при туберкулезе легких.
Mycobacterium tuberculosis является причиной многих заболеваний и смертности во всем мире (1). Растущее число случаев устойчивости к антибиотикам вместе с синергизмом между ВИЧ и туберкулезом усилило наш интерес к этому важному инфекционному заболеванию и к механизмам, способствующим антимикробной защите хозяина.
Микобактерии — это внутриклеточные патогены, которые поглощаются альвеолярными макрофагами (AM) хозяина, 3 , в которых они либо погибают, либо выживают. Выжившие бациллы начинают размножаться и высвобождаться, что приводит к заражению дополнительных клеток-хозяев. Апоптоз AM может быть эффективным оружием для уничтожения или подавления роста внутриклеточных микобактерий. Некоторые данные свидетельствуют о том, что апоптоз AM играет важную роль при туберкулезе. Было показано, что заражение AM человека M. tuberculosis вызывает апоптоз in vitro (2). Кроме того, обширный апоптоз (50–70%) был обнаружен в туберкулезных гранулемах легких больных туберкулезом (2), а значительное увеличение количества апоптотических AM наблюдалось в жидкости бронхоальвеолярного лаважа (BALF) у пациентов с активным туберкулезом легких ( 3, 4). Несмотря на эти наблюдения, неясно, какую роль играет апоптоз AM в патобиологии этого заболевания и увеличивает ли он микобактериальную нагрузку in vivo или снижает ее. Исследования in vitro показывают, что апоптоз может быть механизмом защиты макрофагов от инфекции микобактериями.Действительно, апоптоз человеческих моноцитов ограничивал рост Mycobacterium avium (5), Mycobacterium bovis bacillus Calmette-Guérin (6) и M. tuberculosis (7). Однако исследований in vitro недостаточно для определения чистого влияния истощения AM на реакцию хозяина на туберкулез. AM обладают важными фагоцитарными и иммунными функциями, которые могут быть нарушены процессом апоптоза. Удаление микроорганизмов, которые достигают альвеолярного пространства, частично зависит от фагоцитарных AM.
Кроме того, обширный апоптоз (50–70%) был обнаружен в туберкулезных гранулемах легких больных туберкулезом (2), а значительное увеличение количества апоптотических AM наблюдалось в жидкости бронхоальвеолярного лаважа (BALF) у пациентов с активным туберкулезом легких ( 3, 4). Несмотря на эти наблюдения, неясно, какую роль играет апоптоз AM в патобиологии этого заболевания и увеличивает ли он микобактериальную нагрузку in vivo или снижает ее. Исследования in vitro показывают, что апоптоз может быть механизмом защиты макрофагов от инфекции микобактериями.Действительно, апоптоз человеческих моноцитов ограничивал рост Mycobacterium avium (5), Mycobacterium bovis bacillus Calmette-Guérin (6) и M. tuberculosis (7). Однако исследований in vitro недостаточно для определения чистого влияния истощения AM на реакцию хозяина на туберкулез. AM обладают важными фагоцитарными и иммунными функциями, которые могут быть нарушены процессом апоптоза. Удаление микроорганизмов, которые достигают альвеолярного пространства, частично зависит от фагоцитарных AM. Кроме того, макрофаги представляют микобактериальные АГ CD4 + Т-лимфоцитам, которые играют центральную роль в приобретенной устойчивости к M. tuberculosis . Макрофаги являются важным источником цитокинов 1-го типа при микобактериальной инфекции (8), которые, как известно, важны для развития защитного иммунитета (9). Кроме того, AM продуцируют IFN-γ в ответ на M. tuberculosis (10), который является основным медиатором устойчивости организма к туберкулезу (11, 12). Наконец, мононуклеарные клетки участвуют в образовании гранулем, которые имеют решающее значение для ограничения роста и распространения микобактерий (13).Следовательно, теоретически истощение AM могло иметь положительные и отрицательные эффекты при туберкулезе in vivo.
Кроме того, макрофаги представляют микобактериальные АГ CD4 + Т-лимфоцитам, которые играют центральную роль в приобретенной устойчивости к M. tuberculosis . Макрофаги являются важным источником цитокинов 1-го типа при микобактериальной инфекции (8), которые, как известно, важны для развития защитного иммунитета (9). Кроме того, AM продуцируют IFN-γ в ответ на M. tuberculosis (10), который является основным медиатором устойчивости организма к туберкулезу (11, 12). Наконец, мононуклеарные клетки участвуют в образовании гранулем, которые имеют решающее значение для ограничения роста и распространения микобактерий (13).Следовательно, теоретически истощение AM могло иметь положительные и отрицательные эффекты при туберкулезе in vivo.
В настоящем исследовании мы определили роль истощения AM в инфекции M. tuberculosis у мышей, используя хорошо проверенный метод внутрилегочной доставки инкапсулированного в липосомы дихлорметилендифосфоната (тетрагидрат динатриевой соли клодроновой кислоты, CL 2 МБП). Внутритрахеальное введение инкапсулированного в липосомы CL 2 MBP избирательно истощает AM (14) посредством апоптоза (15, 16), не повреждая другие типы клеток в легком (17).В этой работе мы представляем первые доказательства того, что истощение AM in vivo приводит к улучшенному клиренсу бацилл M. tuberculosis .
Внутритрахеальное введение инкапсулированного в липосомы CL 2 MBP избирательно истощает AM (14) посредством апоптоза (15, 16), не повреждая другие типы клеток в легком (17).В этой работе мы представляем первые доказательства того, что истощение AM in vivo приводит к улучшенному клиренсу бацилл M. tuberculosis .
Материалы и методы
Мыши
Свободных от патогенов 6-недельных самок мышей BALB / c были получены от Harlan Sprague-Dawley (Хорст, Нидерланды) и содержались в помещениях с уровнем биобезопасности 3. Комитет по уходу и использованию животных Амстердамского университета (Амстердам, Нидерланды) одобрил все эксперименты.
Экспериментальная инфекция
Вирулентный лабораторный штамм M. tuberculosis h47Rv выращивали в жидкой среде Дубоша, содержащей 0,01% Твин 80, в течение 4 дней. Повторную культуру инкубировали при 37 ° C, собирали в середине логарифмической фазы и хранили в аликвотах при -70 ° C.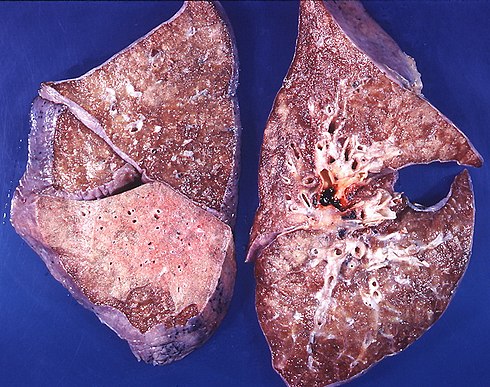 Для каждого эксперимента флакон оттаивали и дважды промывали стерильным 0,9% NaCl. Мышей анестезировали путем ингаляции изофлураном (Abbott Laboratories, Кент, Великобритания) и инфицировали 1 × 10 5 живых бацилл в 50 мкл физиологического раствора, как определено подсчетом жизнеспособных бактерий на чашках с агаром Миддлбрук 7 часов 21 мин.Бактериальное введение проводили интраназально (внутримышечно), как описано ранее (18, 19, 20). Группы из восьми мышей на каждую временную точку были умерщвлены через 2 или 5 недель после инфицирования, и легкие и одна доля печени были удалены в асептических условиях. Органы гомогенизировали с помощью гомогенизатора тканей (Biospec Products, Bartlesville, OK) в 5 объемах стерильного 0,9% NaCl, и 10-кратные серийные разведения высевали на чашки с агаром Миддлбрук 7h21 для определения бактериальной нагрузки. Колонии подсчитывали после 21-дневной инкубации при 37 ° C.Количество КОЕ представлено как общее количество в легких или как общее количество на грамм печени. Для измерения цитокинов гомогенаты легких разбавляли 1/1 в буфере для лизиса (150 мМ NaCl, 15 мМ Трис, 1 мМ MgCl.H 2 O, 1 мМ CaCl 2, 1% Тритон X-100, 100 мкг / мл пепстатина А, лейпептина и апротинина) и инкубировали на льду в течение 30 мин. Супернатанты стерилизовали с использованием фильтра 0,22 мкм (Corning, Corning, NY) и замораживали при -20 ° C до проведения анализов.
Для каждого эксперимента флакон оттаивали и дважды промывали стерильным 0,9% NaCl. Мышей анестезировали путем ингаляции изофлураном (Abbott Laboratories, Кент, Великобритания) и инфицировали 1 × 10 5 живых бацилл в 50 мкл физиологического раствора, как определено подсчетом жизнеспособных бактерий на чашках с агаром Миддлбрук 7 часов 21 мин.Бактериальное введение проводили интраназально (внутримышечно), как описано ранее (18, 19, 20). Группы из восьми мышей на каждую временную точку были умерщвлены через 2 или 5 недель после инфицирования, и легкие и одна доля печени были удалены в асептических условиях. Органы гомогенизировали с помощью гомогенизатора тканей (Biospec Products, Bartlesville, OK) в 5 объемах стерильного 0,9% NaCl, и 10-кратные серийные разведения высевали на чашки с агаром Миддлбрук 7h21 для определения бактериальной нагрузки. Колонии подсчитывали после 21-дневной инкубации при 37 ° C.Количество КОЕ представлено как общее количество в легких или как общее количество на грамм печени. Для измерения цитокинов гомогенаты легких разбавляли 1/1 в буфере для лизиса (150 мМ NaCl, 15 мМ Трис, 1 мМ MgCl.H 2 O, 1 мМ CaCl 2, 1% Тритон X-100, 100 мкг / мл пепстатина А, лейпептина и апротинина) и инкубировали на льду в течение 30 мин. Супернатанты стерилизовали с использованием фильтра 0,22 мкм (Corning, Corning, NY) и замораживали при -20 ° C до проведения анализов.
Истощение AM in vivo
CL 2 MBP был подарком от Roche Diagnostics (Мангейм, Германия).Приготовление липосом, содержащих CL 2 MBP, проводили, как описано ранее (17). Для оценки истощения AM пять неинфицированных мышей в группе находились внутри. инокулировали 100 мкл 0,9% NaCl, липосом PBS или липосом CL 2 MBP. Двумя днями позже AM были количественно определены в BALF. Для экспериментов по туберкулезу 100 мкл физиологического раствора, липосомы PBS или липосомы CL 2 MBP инстиллировали за 2 дня до и через 6, 14 и 25 дней после заражения M. tuberculosis .
Обнаружение апоптотических клеток
Для подтверждения апоптотической гибели клеток, индуцированной липосомами CL 2 MBP, было определено расщепление поли (АДФ-рибоза) полимеразы (PARP), как описано ранее (21). Вкратце, через 7 ч после i.n. инстилляция липосом CL 2 MBP, ткань Tek OTC (Miles Scientific, Naperville, IL) вводили интратрахеально в легкие, которые затем быстро замораживали и хранили при -70 ° C. Срезы криостата (7 мкм) замороженных легких фиксировали в холодном ацетоне в течение 10 мин, инкубировали с 0.3% H 2 O 2 в метаноле в течение 15 минут, блокировали неспецифическое связывание Ig путем инкубации в течение 30 минут с разбавлением 1/10 нормальной козьей сыворотки и инкубировали в течение ночи с сайтом расщепления кроличьими анти-PARP (214 / 215) -специфическое Ab (Biosource International, Camarillo, CA; 5 мкг / мл). После этого следовала 30-минутная инкубация с поли-HRP козьим антикроличьим IgG (Immunovision, Springdale, AZ). Активность пероксидазы выявляли при добавлении субстрата AEC (3-амино-9-этилкарбазол; Sigma, Buchs, Швейцария) и H 2 O 2 .Срезы контрастировали гематоксилином. Отрицательный контроль был создан путем добавления неспецифических контролей изотипа в качестве первичных Abs.
Оценка in vitro действия липосом CL
2 MDP на M. tuberculosis
В общей сложности 2,5 × 10 3 КОЕ инкубировали в восьми повторностях в 96-луночных круглодонных культуральных планшетах в присутствии среды Левенштейна-Йенсена (Becton Dickinson, Franklin Lakes, NJ) либо с 0.9% NaCl, липосомы PBS или липосомы CL 2 MBP. После 48-часовой инкубации при 37 ° C в 5% CO 2 подсчитывали колонии.
Промывание легких
Был проведен бронхоальвеолярный лаваж для получения внутриальвеолярных клеток. Вкратце, мышей анестезировали, обнажали трахею через разрез по средней линии и канюлировали стерильным катетером Abbocath-T 22 размера (Abbott, Sligo, Ирландия). Затем легкие промывали двумя аликвотами по 0,5 мл стерильного раствора 0.9% NaCl. В общей сложности было получено 0,9–1 мл жидкости для лаважа на каждую мышь, и общее количество лейкоцитов определяли с помощью гемацитометра и раствора TÜRK (Merck, Gibbstown, NJ). ЖБАЛ от инфицированных мышей фиксировали 2% параформальдегидом. Количество AM, полиморфно-ядерных клеток (PMN) и лимфоцитов рассчитывали из этих общих значений с использованием препаратов цитоспина, окрашенных модифицированным красителем Гимза (Diff-Quick; Baxter, McGaw Park, IL).
Гистологические анализы
Левое легкое было удалено через 2 или 5 недель после инокуляции M.tuberculosis и фиксировали в 4% параформальдегиде в PBS в течение 24 часов. Одна доля печени неинфицированных мышей была удалена через 2 дня после обработки липосомами CL 2 MBP. После погружения в парафин срезы толщиной 4 мкм окрашивали эозином гематоксилин-эозином или красителем Циля-Нильсена (ZN) на кислотоустойчивые бациллы. Все слайды были закодированы и полуколичественно оценены патологом на общую площадь воспаления (процент поверхности слайда) и формат гранулемы.
Анализ FACS
Клетки легких мышей через 2 и 5 недель после инфицирования (восемь мышей в группе) анализировали с помощью FACS (Becton Dickinson).Суспензию легочных клеток получали с использованием автоматического устройства для дезагрегации (Medimachine System; Dako, Glostrup, Дания) и ресуспендировали в среде (RPMI 1640 (BioWhittaker, Бельгия), 10% FCS, 1% антибиотик-антимикотик (Life Technologies, Rockville, MD)) ). Клетки от двух мышей на группу объединяли для каждой временной точки (с получением четырех образцов для анализа FACS на группу) и доводили до концентрации 4 × 10 6 клеток / мл буфера FACS (добавка PBS с 0,5% BSA, 0,01% NaN 3 и 100 мМ ЭДТА).Иммуноокрашивание молекул клеточной поверхности проводили в течение 30 мин при 4 ° C с использованием непосредственно меченых антител против CD3 (анти-CD3 PE), CD4 (анти-CD4 CyChrome), CD8 (анти-CD8 FITC, анти-CD8 PerCP), CD25 ( анти-CD25 FITC), CD69 (анти-CD69 FITC) и Gr-1 (анти-Gr-1 FITC). Все Abs использовали в концентрациях, рекомендованных производителем (PharMingen, San Diego, CA). Для коррекции неспецифического окрашивания использовали соответствующие контрольные Ab (крысиный IgG2; PharMingen). Клетки фиксировали 2% параформальдегидом, Т-клетки анализировали путем стробирования популяции CD3 + , а гранулоциты — стробированием популяции PMN с прямым и боковым углом рассеяния.Число положительных клеток получали путем установки квадрантного маркера неспецифического окрашивания.
Стимуляция спленоцитов
Суспензии единичных клеток получали измельчением селезенки через клеточный фильтр с размером ячеек 40 мкм (Becton Dickinson). Эритроциты лизировали ледяным изотоническим раствором NH 4 Cl (155 мМ NH 4 Cl, 10 мМ KHCO 3 , 100 мМ EDTA, pH 7,4), а оставшиеся клетки дважды промывали. Спленоциты суспендировали в среде (RPMI 1640 (BioWhittaker), 10% FCS, 1% антибиотик-антимикотик (Life Technologies)), высевали в 96-луночные круглодонные культуральные планшеты при плотности клеток 5 × 10 5 клеток в в трех экземплярах и стимулировали 20 мкг / мл очищенного туберкулином производного белка (PPD; Statens Seruminstitut, Копенгаген, Дания).Супернатанты собирали после 48-часовой инкубации при 37 ° C в 5% CO 2 , и уровни цитокинов анализировали с помощью ELISA.
Анализ пролиферации спленоцитов
Пролиферацию спленоцитов измеряли с помощью теста МТТ, который измеряет восстановление 3- (4,5-диметилтиазол-2-ил) -2,5-дифенилтетразолийбромида до формазана в митохондриях жизнеспособных клеток (22). Спленоциты высевали трижды при плотности 5 × 10 5 клеток / лунку в 96-луночные планшеты с плоским дном и стимулировали 20 мкг / мл PPD.Через 42 часа при 37 ° C в 5% CO 2 клетки инкубировали с 5 мг / мл МТТ (Sigma, Сент-Луис, Миссури) в PBS (pH 7,2) в течение дополнительных 6 часов. Супернатанты декантировали, осадки формазана солюбилизировали добавлением 0,04 н. HCl в изопропаноле и помещали на шейкер для планшетов на 10 мин, после чего клетки растворяли в 2% параформальдегиде. Клеточную пролиферацию количественно оценивали с использованием ридера ELISA при 570 нм. Поглощение необработанных культур было установлено на уровне 100%.
Измерения цитокинов
Цитокины измеряли в гомогенатах легких и супернатантах клеток селезенки с помощью специфических ELISA с использованием согласованных пар антител в соответствии с инструкциями производителя: IFN-γ, IL-2, IL-4 (R&D Systems, Миннеаполис, Миннесота) и IL-10 (PharMingen ).
Статистический анализ
Все значения выражены как среднее ± SEM. Сравнения проводились с тестами Манна-Уитни U . Для сравнения кривых выживаемости использовали анализ Каплана-Мейера с логарифмическим ранговым тестом. Статистически значимыми считались значения p ≤ 0,05.
Результаты
Влияние истощения АМ на течение инфекции
Интраназальное введение инкапсулированного в липосомы CL 2 MBP привело к истощению> 70% AM в ЖБАЛ у неинфицированных мышей через 2 дня (рис.1⇓ а ). Обработка липосомами не повлияла на количество макрофагов. В легких мышей, обработанных липосомами CL 2 MBP, были обнаружены большие области дегенерированных макрофагов с клеточными остатками и апоптотическими тельцами в альвеолярных пространствах. Этот результат согласуется с предыдущими сообщениями о способности вводимых интратрахеально липосом CL 2 MBP или липосом CL 2 MBP, вводимых с помощью аэрозоля, для истощения AM (23, 24). Индукция апоптоза AM обработкой липосомами CL 2 MBP была подтверждена обнаружением расщепленного PARP в легочной ткани (рис.1⇓ б ).
РИСУНОК 1.
a , Влияние липосом CL 2 MBP на AM через 2 дня после внутривенного введения. администрация. Столбики показывают процент общих AM, оставшихся у мышей AM — (▪) или AM + (липосомы) (▨) по сравнению с мышами, получавшими физиологический раствор (□; 100%). Данные представлены как среднее значение и стандартная ошибка среднего для пяти мышей на группу. ∗, p <0,05 AM — мышей против AM + (физиологический раствор) мышей; † , p <0.05 AM — мышей против AM + (липосомы) мышей. b , Апоптотические AMs визуализировали путем иммуноокрашивания ткани легких на расщепленный PARP через 7 часов после обработки липосомами CL 2 MBP (исходное увеличение, × 80).
Чтобы исследовать роль AM в исходе туберкулеза, CL 2 липосомы MBP вводили внутримышечно. 2 дня до и 6, 14 и 25 дней после индукции туберкулеза (AM — мышей). Мыши, получавшие физиологический раствор или липосомы, служили контролем (мыши AM + ).Выживаемость и бактериальная нагрузка в легких и печени были проанализированы для определения устойчивости к туберкулезу. Как показано на фиг. 2⇓, выживаемость мышей AM + (физиологический раствор) значительно снизилась с 35 дня и далее, что привело к 90% смертности через 5 месяцев. В отличие от них, все мыши AM — контролировали одну и ту же инфекционную дозу и пережили 5-месячное наблюдение ( p <0,0001). Кроме того, у мышей AM + (липосомы) было обнаружено значительное снижение выживаемости по сравнению с животными AM — ( p = 0.029).
ФИГУРА 2.
Влияние истощения AM на выживаемость мышей после инфицирования M. tuberculosis . Мышей BALB / c ( n = 10 на группу) вводили внутримышечно. вводили с физиологическим раствором, липосомами или липосомами CL 2 MBP до и после бактериального заражения 1 × 10 5 M. tuberculosis h47Rv. ∗, p <0,05 AM — мышей против AM + (физиологический раствор) мышей; † , p <0.05 AM — мыши против мышей AM + (липосомы); ‡ , p
Из-за впечатляющих различий в выживаемости мы определили, существуют ли различия в микобактериальной нагрузке на ранних этапах инфекции. Количество бактерий, отложившихся в легких и печени, определяли через 1 день после заражения. Количество бактерий в легких было сопоставимо с количеством бактерий, которым вводили интраназально, и не различалось между группами.Количество M. tuberculosis КОЕ, извлеченных из легких, существенно не различались между AM + (физиологический раствор / липосомы) и AM — мышей через 2 недели после инфицирования (рис. 3⇓ a ). Через 5 недель после инфицирования значимые различия в содержании тканей бацилл M. tuberculosis наблюдались у мышей AM + и AM —. Легкие мышей AM — содержали в 9,7 раз меньше жизнеспособных микобактерий, чем легкие животных AM + (физиологический раствор) ( p = 0.021, рис. 3⇓ а ) и в 7,1 раза меньше, чем у мышей AM + (липосомы) ( p = 0,035).
РИСУНОК 3.
Рост M. tuberculosis в легких ( a ) и печени ( b ) мышей AM + (физиологический раствор / липосомы) и мышей AM — . Данные представляют собой средние значения и SEM для КОЕ от восьми мышей на группу для каждой временной точки. ∗, p <0,05 AM — мышей против AM + (физиологический раствор) мышей; †, p <0.05 AM — мышей против AM + (липосомы) мышей.
Для большей ясности в отношении места обитания микобактерий у животных AM — промывные жидкости этих мышей анализировали с помощью красителя Циля-Нильсена (ZN) на кислотоустойчивые бациллы. В ЖБАЛ мышей AM + микобактерии присутствовали в цитоплазме в 39 ± 4% (через 2 недели после инфицирования) и 47 ± 10% (через 5 недель после заражения) макрофагов и в нескольких PMN. В ЖБАЛ мышей AM — мы обнаружили внеклеточные микобактерии, но микобактерии особенно присутствовали в клеточных остатках.
Через 2 недели после инфицирования микобактериальная нагрузка в печени мышей AM — была увеличена по сравнению с мышами AM + (физиологический раствор) ( p = 0,02) и AM + (липосомы) мышами ( p = 0,004, рис.3 ⇑ б ). Через 5 недель после инфицирования количество организмов в печени мышей AM — было в 3,6 раза меньше, чем у мышей AM + (физиологический раствор) ( p = 0,011, рис. 3⇑ b ) и 2,7 раз ниже, чем у AM + (липосомы) животных (существенно не отличается).Количество бактерий в легких и печени контрольных групп мышей, получавших физиологический раствор или липосомы, существенно не различались в любой момент времени.
Чтобы исключить возможность того, что инкапсулированный в липосомы CL 2 MBP имел прямое действие на микобактерии, M. tuberculosis инкубировали in vitro в присутствии или в отсутствие этого агента в течение 2 дней. Подсчет бактерий не показал прямого антимикобактериального эффекта инкапсулированного в липосомы CL 2 MBP (данные не показаны).
В совокупности эти данные предполагают, что истощение AM в результате апоптоза может играть важную роль в борьбе с инфекцией M. tuberculosis .
Гистология
Через две недели после инокуляции M. tuberculosis в легких мышей AM + (физиологический раствор / липосомы) наблюдались более или менее четко выраженные гранулемы, состоящие из большей части эпителиоидных и пенистых клеток и небольшого количества лимфоцитов по всей паренхиме (рис. .4⇓ а ). Плотные лимфоцитарные инфильтраты также присутствовали вокруг мелких сосудов. В легких мышей AM — обнаружен относительно диффузный инфильтрат гранулоцитов с заметными периваскулярными лимфоцитарными инфильтратами. Четко выраженных гранулем не было (рис. 4⇓ b ). Процент воспаленной паренхимы был одинаковым во всех группах (мыши AM + (физиологический раствор), 21,25 ± 3,5%; мыши AM + (липосомы), 16,9 ± 3,5%; мыши AM — , 18,8 ± 2,9%). .
РИСУНОК 4.
Гистопатология легких. a , через две недели после инфицирования, в легких мышей AM + были обнаружены гранулемы правильной формы, в основном состоящие из макрофагов и небольшого количества лимфоцитов (окраска гематоксилином и эозином; исходное увеличение × 50). b , В этот же момент в легких мышей AM — обнаружены незначительные периваскулярные лимфоцитарные инфильтраты и дегенерированные макрофаги в альвеолярных пространствах. Четко выраженных гранулем не обнаружено (окраска гематоксилином и эозином; исходное увеличение × 50). c , через пять недель после инфицирования, в легких мышей AM + обнаружен диффузный воспалительный инфильтрат, преимущественно состоящий из макрофагов (окраска гематоксилином и эозином; исходное увеличение × 25). d , Легкие мышей AM — имели почти нормальный вид (окрашивание гематоксилином и эозином; исходное увеличение × 25). Показанные слайды являются репрезентативными для восьми мышей в группе для каждой временной точки.
Через 5 недель воспалительные инфильтраты в легких у всех мышей стали более диффузными и интенсивными с клеточным составом, сопоставимым у мышей AM + (физиологический раствор / липосомы) и AM —.Однако процент воспаленной паренхимы был меньше у мышей AM — (45 ± 4,8%), чем у мышей AM + (физиологический раствор, 61,9 ± 4,9%; липосомы, 62,9 ± 7,1%) (рис. 4⇑, ). c и d ).
Подмножества ячеек
CD4 + Т-клетки играют важную роль в защитном иммунитете против инфекции M. tuberculosis (25, 26, 27) и должны стимулироваться специфическими лигандами на поверхности APC. Чтобы изучить, важны ли AM для индукции CD4 + Т-клеточного иммунитета, мы исследовали фенотипы иммунных клеток в легких с помощью анализа FACS.Как показано в Таблице I the, процентное содержание Т-клеток CD4 + не различалось у мышей AM + и AM — через 5 недель после заражения и было немного снижено у мышей AM — через 2 недели после заражения. Через две недели после заражения было продемонстрировано, что лимфоциты CD4 + мышей AM — были более активными, чем лимфоциты CD4 + мышей AM + , согласно оценке маркеров активации CD69 и CD25.
Таблица I.
Влияние истощения AM на клеточные субпопуляции в легких при туберкулезе a
Предполагается, что помимо CD4 + T-клеток, CD8 + T-клетки также участвуют в защите хозяина от микобактериальных инфекций (25).Процент CD8 + Т-клеток в гомогенатах легких не изменился у мышей AM — по сравнению с мышами AM + (физиологический раствор / липосомы) через 5 недель после инфицирования и немного увеличился через 2 недели после заражения. Экспрессия маркера активации CD69 на этих клетках была немного увеличена. Экспрессию CD25 на клетках CD8 + обнаружить не удалось. Абсолютное количество PMN было выше у мышей AM — , чем у контрольных животных AM + через 2 недели после инфицирования.Через 5 недель количество общих лейкоцитов снизилось у мышей AM — по сравнению с мышами AM + (физиологический раствор / липосомы), что, вероятно, отражает тяжесть заболевания.
Чтобы получить более полное представление о притоке лейкоцитов в альвеолярный компартмент, легкие промывали, клетки подсчитывали и препараты цитоспина окрашивали эозином гематоксилин-эозином (таблица II). Через две недели после инфицирования количество лейкоцитов было выше у мышей AM — , чем у мышей AM + (физиологический раствор / липосомы).В соответствии с количеством лейкоцитов в легких через 5 недель после инфицирования, количество клеток в ЖБАЛ было ниже у мышей AM —, чем у мышей AM + (физиологический раствор / липосомы). Как и следовало ожидать, животные, получавшие липосомы CL 2 , имели в 2 раза меньше AM в их ЖБАЛ, чем мыши AM + (физиологический раствор / липосомы). Однако количество PMN и лимфоцитов у мышей AM — было увеличено через 2 недели после инфицирования по сравнению с мышами AM + (физиологический раствор / липосомы).Вследствие более низкого количества лейкоцитов у мышей AM — через 5 недель после инфицирования количество PMN и лимфоцитов было снижено в этой группе по сравнению с мышами AM + (физиологический раствор / липосомы).
Таблица II.
Влияние истощения AM на клеточный состав ЖБАЛ при туберкулезе a
Паттерны экспрессии цитокинов в легких
Поскольку развитие раннего клеточного иммунитета Th2 необходимо для устранения M.tuberculosis (9), мы исследовали, был ли улучшенный исход туберкулеза, наблюдаемый у мышей AM —, связан со сдвигом продукции цитокинов на ранней стадии инфекции. Поэтому мы измерили концентрации цитокинов Th2 (IFN-γ и IL-2) и Th3 (IL-4, IL-10) в легких. Как показано на фиг. 5⇓, все цитокины были снижены у мышей AM — по сравнению с мышами AM + (физиологический раствор / липосомы) через 2 недели после заражения. Важно отметить, что по сравнению с мышами AM + (физиологический раствор / липосомы) концентрации цитокинов Th3 были относительно более низкими, чем уровни цитокинов Th2 у мышей AM —.Как следствие, более глубокий ответ Th2 был обнаружен в легких мышей AM —.
РИСУНОК 5.
Влияние истощения AM на M. tuberculosis -опосредованную индукцию цитокинов 1 типа ( a ) и 2 типа ( b ) в легких мышей AM + (физиологический раствор) (□), AM + (липосомы) мышей (▨) и AM — мышей (▪) через 2 недели после инфицирования. Данные представляют собой среднее значение и стандартную ошибку среднего для восьми мышей. ∗, п <0.05 AM — мыши против мышей AM + (физиологический раствор); †, p <0,05 AM — мышей против AM + (липосомы) мышей.
Цитокин и пролиферативный ответ стимулированных ex vivo клеток селезенки
Способность клеток селезенки, собранных через 2 недели после инфицирования M. tuberculosis , продуцировать цитокины ex vivo при стимуляции PPD, была исследована как еще одна мера ответа Th2 по сравнению с Th3. Клетки селезенки мышей AM — секретировали 3.Уровни IFN-γ в 5 раз выше, чем в спленоцитах мышей AM + (физиологический раствор), и в 2 раза выше, чем в спленоцитах мышей AM + (липосомы) (фиг. 6⇓). ИЛ-4 не обнаруживался в супернатантах спленоцитов, стимулированных PPD, во всех группах. При стимуляции покрытыми анти-CD3 и анти-CD28 АТ спленоциты мышей AM — секретировали более высокие уровни IFN-γ и значительно более низкие уровни IL-4 по сравнению с животными AM + (физиологический раствор). Кроме того, реакции пролиферации спленоцитов на PPD оценивали с помощью анализа включения MTT.Мы обнаружили, что спленоциты мышей AM — индуцировали самый сильный пролиферативный ответ на PPD, хотя и не статистически значимый (мыши AM + (физиологический раствор), 155 ± 17%; мыши AM + (липосомы), 158 ± 6%). ; AM — мышей, 195 ± 31%).
РИСУНОК 6.
Спленоциты инфицированных мышей AM — (▪) выделяют больше IFN-γ в ответ на PPD и анти-CD3 / 28 Abs и меньше IL-4 в ответ на анти-CD3 / 28 ABS, чем спленоциты инфицированных AM + мыши (□, физиологический раствор; ▨, липосомы).Спленоциты собирали через 2 недели после внутривенного введения. инокуляция M. tuberculosis и стимуляция в течение 48 часов. Данные представляют собой среднее значение и стандартную ошибку среднего для восьми мышей на группу. ∗, p <0,05 AM — мышей против AM + (физиологический раствор) мышей; †, p <0,05 AM — мышей против AM + (липосомы) мышей; ‡, p
Обсуждение
AM
могут играть двойную роль во время заражения M.туберкулез . С одной стороны, у AM есть несколько инструментов для борьбы с внутриклеточными патогенами, такими как продукция IFN-γ и токсичных эффекторных молекул (реактивные промежуточные соединения кислорода и реактивные промежуточные соединения азота), а также лишение доступности внутриклеточного железа. С другой стороны, микобактерии могут частично полагаться на внутриклеточную среду AM для их размножения. В этом исследовании мы демонстрируем, что истощение AM in vivo улучшает исход туберкулеза легких, на что указывает полная защита от летального исхода и замедление роста микобактерий в легких.Эти результаты предполагают, что AM способствуют росту M. tuberculosis в легочном компартменте и что апоптоз AM может быть частью механизмов защиты хозяина во время туберкулеза. Интересно, что AM, по-видимому, действительно играют важную роль в начальном улавливании микобактерий, о чем свидетельствует наблюдение, что через 2 недели после инфицирования мыши AM — имели больше микобактерий в печени.
Апоптоз клетки-хозяина уже показал себя как защитную стратегию для ограничения роста вирусов, которые, как и микобактерии, живут внутриклеточно (28, 29, 30).Тот факт, что апоптоз AM может способствовать защите хозяина, дополнительно подтверждается наблюдениями за обратной зависимостью между апоптозом и вирулентностью, т. Е. Вирулентный штамм M. tuberculosis h47Rv индуцировал меньше апоптоза при инфицировании AM человека, чем аттенуированный штамм h47Ra (2). . Следовательно, микобактерии, по-видимому, разработали способы модуляции защитного апоптотического процесса AM, и индуцированное патогеном подавление пути гибели клетки-хозяина может служить для уклонения от защиты хозяина, которая может действовать, чтобы ограничить инфекцию.Следует отметить, что роль АМ в респираторных инфекциях, вызываемых внеклеточно растущими патогенами, противоположна. Действительно, индукция апоптоза AM во время Klebsiella pneumonia нарушает механизмы защиты хозяина (31).
Самая прямая интерпретация улучшенного исхода туберкулеза у мышей AM — заключается в том, что истощение AM снижает жизнеспособность M. tuberculosis , потому что среда для внутриклеточной репликации и сокрытия разрушается (32).Кроме того, апоптозные тельца поддерживают целостность своей плазматической мембраны, так что бациллы содержатся из внеклеточной среды и могут быть поглощены вновь привлеченными AM (5). Дополнительным объяснением защиты, наблюдаемой при истощении AM, может быть то, что в раннем иммунном ответе доминировал профиль Th2-типа, который важен для устойчивости к микобактериям (9). Преобладание цитокинов Th2-типа у мышей AM — существовало как в легочной ткани, в которой особенно снижены концентрации цитокинов Th3, так и в супернатантах спленоцитов, стимулированных PPD.Wang et al. (8) недавно сообщили, что макрофаги легких, собранные во время микобактериальной инфекции, выделяют значительное количество цитокинов 1 типа. В соответствии с этими наблюдениями мы обнаружили более низкие уровни IFN-γ и IL-2 в гомогенатах легких через 2 недели после инфицирования. Однако чистым эффектом истощения АМ in vivo было относительное преобладание 1-го типа в легких. Маловероятно, что этот сдвиг типа 1 был следствием более мягкой воспалительной реакции у мышей AM –, которые, во всяком случае, демонстрировали немного больше признаков воспаления (таблицы I⇑ и II⇑).Вполне возможно, что в этом сдвиге было задействовано истощение AM. AM являются типичными популяциями макрофагов, которые, как известно, индуцируют дифференцировку наивных Т-клеток в клетки типа Th3 и проявляют связанные с Th3 эффекторные функции (33, 34). Кроме того, разумно, что в этот сдвиг вовлечены пониженные уровни IL-10 у мышей AM —, учитывая, что IL-10 происходит из AM (35, 36) и что этот цитокин подавляет продукцию цитокинов типа 1 (37). ). Другой причиной лучшего результата у мышей AM — может быть усиленный приток PMN и активированных лимфоцитов в легкие мышей AM — (таблицы I⇑ и II⇑).Т-клетки играют важную роль в защитном иммунитете против M. tuberculosis (38), и, следовательно, повышенное количество этих клеток, скорее всего, способствует устойчивости. В подтверждение роли PMN в микобактериальных инфекциях, защитная функция нейтрофильной реакции была продемонстрирована повышенной чувствительностью мышей, лишенных нейтрофилов, к M. tuberculosis (39). Вполне возможно, что при отсутствии фагоцитирующих AM у мышей AM — PMN получают больше сигналов для миграции в легкие для поглощения и удаления апоптотических тел, полученных из AM.
AM — мыши были полностью или даже более способны к привлечению и активации Т-клеток в легочном отделе. В этом контексте следует отметить, что AM являются плохими APC (40, 41, 42, 43) и что дендритные клетки (44, 45) и интерстициальные макрофаги (46) считаются наиболее эффективными APC в легких. Эксперименты in vitro даже указывают на то, что AM обладают высокой супрессией Т-клеток (47, 48, 49). Вполне возможно, что подавляющие эффекты AM на легочный иммунный ответ могут служить для ограничения повреждений, вызванных тяжелыми иммунными ответами в легочной ткани, но в то же время могут ослаблять защиту хозяина во время туберкулеза.
Через две недели после инфицирования из печени мышей AM – было выделено на КОЕ M. tuberculosis, чем из печени мышей AM + . Наше исследование не проясняет механизмы, способствующие этому наблюдению. Возможно, бациллы, которые остаются внеклеточно (например, у мышей AM –), легче получают доступ к кровообращению и лимфатическому кровотоку. Маловероятно, что ингаляция липосом CL 2 MBP влияла на количество и / или функцию клеток Купфера в печени, учитывая, что липосомы не способны преодолевать стенки капилляров и другие сосудистые барьеры (17).Отсутствие эффекта ингаляционных липосом CL 2 MBP на клетки Купфера подтверждается тем фактом, что гистология печени не различалась между разными группами лечения (данные не показаны).
Липосомы использовали для инкапсулирования Cl 2 MBP, потому что эти фосфолипидные сферы активно поглощаются макрофагами. Липосомы могут снижать фагоцитарное и миграционное поведение AM (50) и, следовательно, могут влиять на защиту хозяина от M. tuberculosis . В соответствии с этим животные, получавшие i.п. только с липосомами (т.е. без CL 2 MBP), показали повышенную выживаемость и небольшое (незначительное) снижение M. tuberculosis КОЕ в легких и печени по сравнению с мышами AM + (физиологический раствор). Так как мы стремились определить роль AM в туберкулезе легких, контрольные мыши должны иметь нормальные, не подавленные AM (17), и таким образом мы считаем животных AM + (физиологический раствор) лучше контрольными, чем мышей AM + (липосомы). . Физическое истощение AM липосомами CL 2 MBP гарантирует, что все функции макрофагов, которые проглотили это соединение, отменяются.Ожидается, что AM, которые фагоцитируют только липосомы, будут иметь некоторые функциональные нарушения. Тем не менее, мыши, получавшие липосомы AM + , значительно отличались от мышей AM — в отношении всех измеренных ответов.
Это исследование является первым, показывающим, что истощение AM in vivo защищает от инфекции M. tuberculosis и что оно связано с усилением Th2-опосредованного иммунного ответа. Таким образом, апоптоз AM, наблюдаемый у больных туберкулезом, может быть важным процессом защиты от микобактерий.Настоящие результаты не только дают новое понимание возможных механизмов противомикробной защиты макрофагов, но также раскрывают потенциально новые терапевтические стратегии для лечения внутриклеточных бактериальных заболеваний.
Благодарности
Мы благодарим Joost Daalhuisen, Nike Claessen и доктора Arend Kolk за квалифицированную техническую помощь.
Сноски
№1 Работа поддержана грантом Нидерландской организации научных исследований (J.C.L.) и стипендию Королевской голландской академии искусств и наук (T.v.d.P.).
№2 Направляйте запросы на переписку и перепечатку доктору Яклиену Лимансу, Лаборатория экспериментальной внутренней медицины, Академический медицинский центр, Амстердамский университет, G2-105, Meibergdreef 9, 1105 AZ Амстердам, Нидерланды. Адрес электронной почты: j.c.leemans {at} amc.uva.nl
№3 В статье использованы сокращения: AM — альвеолярный макрофаг; BALF, жидкость бронхоальвеолярного лаважа; Cl 2 MBP, дихлорметиленбисфосфонат; я.п., интраназальный; PARP, поли (АДФ-рибоза) полимераза; PMN, полиморфно-ядерная клетка; PPD, очищенное производное белка.
- Получено 14 апреля 2000 г.
- Принято 22 января 2001 г.
- Copyright © 2001 Американская ассоциация иммунологов
Ссылки
- 45
Dye, C., S. Scheele, P. Dolin, V. Pathania, M. C. Raviglione. 1999. Заявление о консенсусе: глобальное бремя туберкулеза: оценочная заболеваемость, распространенность и смертность по странам: Глобальный проект ВОЗ по эпиднадзору и мониторингу.Варенье. Med. Доц. 282: 677
- ↵
Кин, Дж., М. К. Бальцевич-Саблинска, Х. Г. Ремольд, Г. Л. Чупп, Б. Б. Мик, М. Дж. Фентон, Х. Корнфельд. 1997. Инфекция Mycobacterium tuberculosis способствует апоптозу альвеолярных макрофагов человека. Заразить. Иммун. 65: 298
- ↵
Klingler, K., K. M. Tchou-Wong, O. Brandli, C. Aston, R. Kim, C. Chi, W. N. Rom. 1997. Влияние микобактерий на регуляцию апоптоза в мононуклеарных фагоцитах.Заразить. Иммун. 65: 5272
- ↵
Пласидо Р., Дж. Манчино, А. Амендола, Ф. Мариани, С. Вендетти, М. Пьячентини, А. Сандуцци, М. Л. Боккино, М. Зембала, В. Колицци. 1997. Апоптоз человеческих моноцитов / макрофагов при инфекции Mycobacterium tuberculosis . J. Pathol. 181: 31
- ↵
Fratazzi, C., R. D. Arbeit, C. Carini, H. G. Remold. 1997. Запрограммированная гибель клеток макрофагов человека, инфицированных Mycobacterium avium сероваром 4, предотвращает распространение микобактерий и вызывает ингибирование роста микобактерий за счет недавно добавленных неинфицированных макрофагов.J. Immunol. 158: 4320
- ↵
Моллой А., Лаочумроонворапонг П., Каплан Г. 1994. Апоптоз, но не некроз инфицированных моноцитов сочетается с уничтожением внутриклеточной палочки Кальметта-Герена. J. Exp. Med. 180: 1499
- ↵
Оддо, М., Т. Ренно, А. Аттингер, Т. Баккер, Х. Р. Макдональд, П. Р. Мейлан. 1998. Апоптоз инфицированных человеческих макрофагов, индуцированный Fas-лигандом, снижает жизнеспособность внутриклеточных Mycobacterium tuberculosis .J. Immunol. 160: 5448
- ↵
Ван Дж., Дж. Уэйкхэм, Р. Харкнесс, З. Син. 1999. Макрофаги являются важным источником цитокинов 1-го типа при микобактериальной инфекции. J. Clin. Вкладывать деньги. 103: 1023
- ↵
Рук, Г. А., Р. Эрнандес-Пандо. 1996. Патогенез туберкулеза. Анну. Rev. Microbiol. 50: 259
- ↵
Фентон, М.J., M. W. Vermeulen, S. Kim, M. Burdick, R.M Strieter, H. Kornfeld. 1997. Индукция продукции γ-интерферона в альвеолярных макрофагах человека Mycobacterium tuberculosis . Заразить. Иммун. 65: 5149
- ↵
Флинн, Дж. Л., Дж. Чан, К. Дж. Трибольд, Д. К. Далтон, Т. А. Стюарт, Б. Р. Блум. 1993. Существенная роль интерферона γ в устойчивости к инфекции Mycobacterium tuberculosis . J. Exp. Med. 178: 2249
- ↵
Купер, А.М., Д. К. Далтон, Т. А. Стюарт, Дж. П. Гриффин, Д. Г. Рассел, И. М. Орм. 1993. Диссеминированный туберкулез у мышей с нарушенным геном интерферона γ. J. Exp. Med. 178: 2243
- ↵
Чан, Дж., С. Х. Э. Кауфманн. 1994. Иммунные механизмы защиты. Б. Р. Блум, изд. Туберкулез: патогенез, защита и контроль 389-415. ASM Press, Вашингтон, округ Колумбия
- ↵
Тепен, Т., Н. Ван Ройен, Г. Крааль. 1989. Элиминация альвеолярных макрофагов in vivo связана с усилением легочного иммунного ответа у мышей. J. Exp. Med. 170: 499
- ↵
Наито М., Х. Нагаи, С. Кавано, Х. Умедзу, Х. Чжу, Х. Морияма, Т. Ямамото, Х. Такацука, Ю. Такеи. 1996. Инкапсулированный в липосомы дихлорметилендифосфонат вызывает апоптоз макрофагов in vivo и in vitro. J. Leukocyte Biol. 60: 337
- ↵
Ван Ройен, Н., А. Сандерс, Т. К. ван ден Берг. 1996. Апоптоз макрофагов, индуцированный липосомной внутриклеточной доставкой клодроната и пропамидина. J. Immunol. Методы 193: 93
- ↵
Ван Ройен, Н., А. Сандерс. 1994. Истощение макрофагов, опосредованное липосомами: механизм действия, приготовление липосом и приложения. J. Immunol. Методы 174: 83
- ↵
Сондерс, Б.М., К. Ура. 1996. Интраназальное инфицирование бежевых мышей комплексом Mycobacterium avium : роль нейтрофилов и естественных клеток-киллеров. Заразить. Иммун. 64: 4236
- ↵
Эрб, К. Дж., Дж. У. Холлоуэй, А. Собек, Х. Молл, Г. Ле Гро. 1998. Заражение мышей Mycobacterium bovis -bacillus Calmette-Guérin (BCG) подавляет вызванную аллергеном эозинофилию дыхательных путей. J. Exp. Med. 187: 561
- ↵
Юфферманс, Н.П., С. Флоркин, Л. Камольо, А. Вербон, А. Х. Колк, П. Спилман, С. Дж. Ван Девентер, Т. ван дер Пол. 2000. Передача сигналов интерлейкина-1 важна для защиты хозяина во время туберкулеза легких у мышей. J. Infect. Dis. 182: 902
- ↵
Duriez, P. J., G. M. Shah. 1997. Расщепление поли (АДФ-рибозы) полимеразы: чувствительный параметр для изучения гибели клеток. Biochem. Cell Biol. 75: 337
- ↵
Джордан, Дж.П., К. М. Хэнд, Р. С. Марковиц, П. Блэк. 1992. Тест на химиотерапевтическую чувствительность церебральных глиом: использование колориметрического теста МТТ. J. Neurooncol. 14: 19
- ↵
Berg, J. T., S. T. Lee, T. Thepen, C. Y. Lee, M. F. Tsan. 1993. Истощение альвеолярных макрофагов инкапсулированным в липосомы дихлорметилендифосфонатом. J. Appl. Physiol. 74: 2812
- ↵
Коогучи, К., С. Хашимото, А. Кобаяси, Ю. Китамура, И. Кудох, Дж. Винер-Крониш, Т. Сава. 1998. Роль альвеолярных макрофагов в инициации и регуляции воспаления при пневмонии Pseudomonas aeruginosa . Заразить. Иммун. 66: 3164
- ↵
Купер А. М., Дж. Л. Флинн. 1995. Защитный иммунный ответ на Mycobacterium tuberculosis . Curr. Мнение. Иммунол. 7: 512
- ↵
Андерсен, П.. 1997. Ответы хозяев и антигены, участвующие в защитном иммунитете к Mycobacterium tuberculosis . Сканд. J. Immunol. 45: 115
- ↵
Карузо, А. М., Н. Сербина, Э. Кляйн, К. Трибольд, Б. Р. Блум, Дж. Л. Флинн. 1999. Мыши, дефицитные по CD4 Т-клеткам, имеют лишь временно сниженные уровни IFN-γ, но умирают от туберкулеза. J. Immunol. 162: 5407
- ↵
Антони, Б.А., П. Саббатини, А. Б. Рабсон, Э. Уайт. 1995. Ингибирование апоптоза в клетках, инфицированных вирусом иммунодефицита человека, увеличивает выработку вируса и способствует хронической инфекции. J. Virol. 69: 2384
- ↵
Vaux, D. L., G. Haecker, A. Strasser. 1994. Эволюционная перспектива апоптоза. Ячейка 76: 777
- ↵
Клем, Р. Дж., Л. К. Миллер. 1993. Апоптоз снижает как репликацию in vitro, так и инфекционность in vivo бакуловируса.J. Virol. 67: 3730
- ↵
Broug-Holub, E., G. B. Toews, J. F. van Iwaarden, R. M. Strieter, S. L. Kunkel, R. Paine, III, T. J. Standiford. 1997. Альвеолярные макрофаги необходимы для защитной защиты легких у мышей Klebsiella пневмонии: удаление альвеолярных макрофагов увеличивает рекрутирование нейтрофилов, но снижает бактериальный клиренс и выживаемость. Заразить. Иммун. 65: 1139
- ↵
Оттенхофф, Т.Х., Мутис Т. 1995. Роль цитотоксических клеток в защитном иммунитете и иммунопатологии внутриклеточных инфекций. Евро. J. Clin. Вкладывать деньги. 25: 371
- ↵
Cua, D. J., S. A. Stohlman. 1997. Эффекты in vivo цитокинов Т-хелперов типа 2 на индукцию субпопуляций Т-хелперов макрофагальными антиген-представляющими клетками. J. Immunol. 159: 5834
- ↵
Goerdt, S., C.E. Orfanos.1999. Другие функции, другие гены: альтернативная активация антигенпрезентирующих клеток. Иммунитет 10: 137
- ↵
Мартинес, Дж. А., Т. Э. Кинг, мл., К. Браун, К. А. Дженнингс, Л. Бориш, Р. Л. Мортенсон, Т. З. Хан, Т. В. Бост, Д. В. Ричес. 1997. Повышенная экспрессия гена интерлейкина-10 альвеолярными макрофагами при интерстициальном заболевании легких. Являюсь. J. Physiol. 273: L676
- ↵
Маньян, А., Д. ван Пи, П. Бонгранд, Д. Вервлоэт. 1998. Продукция интерлейкина (IL) -10 и IL-12 альвеолярных макрофагов при атопической астме. Аллергия 53: 1092
- ↵
Мосманн Т. Р. 1994. Свойства и функции интерлейкина-10. Adv. Иммунол. 56: 1
- ↵
Stenger, S., R.L. Modlin. 1999. Т-клеточный иммунитет против Mycobacterium tuberculosis . Curr. Мнение. Microbiol. 2: 89
- ↵
Педроса, Дж., Б. М. Сондерс, Р. Аппельберг, И. М. Орм, М. Т. Сильва, А. М. Купер. 2000. Нейтрофилы играют защитную нефагоцитарную роль при системном инфицировании мышей Mycobacterium tuberculosis . Заразить. Иммун. 68: 577
- ↵
Toews, G. B., W. C. Vial, M. M. Dunn, P. Guzzetta, G. Nunez, P. Stastny, M. F. Lipscomb. 1984. Функция дополнительных клеток альвеолярных макрофагов человека в специфической пролиферации Т-клеток. J. Immunol. 132: 181
- ↵
Липскомб, М.Ф., К. Р. Лайонс, Г. Нуньес, Э. Дж. Болл, П. Стастны, В. Виал, В. Лем, Дж. Вайслер, Л. М. Миллер. 1986. Альвеолярные макрофаги человека: HLA-DR-положительные макрофаги, которые являются слабыми стимуляторами первичной реакции смешанных лейкоцитов. J. Immunol. 136: 497
- ↵
Lyons, C. R., E. J. Ball, G. B. Toews, J. C. Weissler, P. Stastny, M. F. Lipscomb. 1986. Неспособность альвеолярных макрофагов человека стимулировать покоящиеся Т-клетки коррелирует со снижением связывания антиген-специфических Т-клеток с макрофагами.J. Immunol. 137: 1173
- ↵
Крадин, Р. Л., К. М. Маккарти, К. И. Дейли, А. Бердшоу, Дж. Т. Курник, Э. Э. Шнибергер. 1987. Плохая функция дополнительных клеток макрофагов у крыс может отражать их неспособность образовывать кластеры с Т-клетками. Clin. Иммунол. Immunopathol. 44: 348
- ↵
Сертл, К., Т. Такемура, Э. Чахлер, В. Дж. Ферранс, М. А. Калинер, Э. М. Шевач.1986. Дендритные клетки с антигенпрезентирующей способностью находятся в эпителии дыхательных путей, паренхиме легких и висцеральной плевре. J. Exp. Med. 163: 436
- ↵
Masten, B.J., M. F. Lipscomb. 1999. Сравнение дендритных клеток легких и В-клеток в стимуляции наивных антиген-специфических Т-клеток. J. Immunol. 162: 1310
- ↵
Franke-Ullmann, G., C. Pfortner, P. Walter, C. Steinmuller, M.Л. Ломанн-Маттес, Л. Кобзик. 1996. Характеристика интерстициальных макрофагов легких мышей в сравнении с альвеолярными макрофагами in vitro. J. Immunol. 157: 3097
- ↵
Холт П. Г. 1979. Альвеолярные макрофаги. II. Подавление пролиферации лимфоцитов очищенными макрофагами из легких крысы. Иммунология 37: 429
- ↵
Маккомбс, К. К., Дж. П. Михальский, Б. Т. Вестерфилд, Р.W. Light. 1982. Альвеолярные макрофаги человека подавляют пролиферативную реакцию лимфоцитов периферической крови. Сундук 82: 266
- ↵
Билык Н., Холт П.Г. 1995. Цитокиновая модуляция иммуносупрессивного фенотипа популяций легочных альвеолярных макрофагов. Иммунология 86: 231
- ↵
Де Хаан, А., Г. Гроен, Дж. Проп, Н. ван Ройен, Дж. Вильшут. 1996. Иммуноадъювантная активность липосом слизистой оболочки: роль альвеолярных макрофагов.Иммунология 89: 488
ТБ Онлайн — ТБ легких
Туберкулез легких определяется как активная инфекция легких (латинское pulmo = легкое). Это наиболее серьезная инфекция туберкулеза, потому что инфекция легких очень заразна из-за способа передачи через капельку. Это может быть опасно для жизни пациента: при отсутствии лечения более 50% пациентов с туберкулезом легких умирают. Во всем мире 87% всех случаев туберкулеза, зарегистрированных в 2004 г., были либо только туберкулезом легких, либо включали туберкулез легких.
Рентген показывает легочную туберкулезную инфекцию
Большинство случаев туберкулеза легких возникают после первичной инфекции. Это означает, что после заживления первоначальной первичной инфекции гранулема (масса иммунных клеток, окружающих туберкулезную инфекцию, предотвращающую ее дальнейшее повреждение), которая образовалась во время этого процесса, по-прежнему содержит бактерии туберкулеза, которые могут выживать годами (см. Туберкулез поражает организм).
Если иммунная система человека с гранулемой ТБ ухудшается, эти бактерии могут реактивироваться, и ТБ может снова вспыхнуть. Как только бациллы туберкулеза реактивируются, они быстро разрушают легочную ткань вокруг гранулемы. Это вызывает серьезное повреждение ткани, которая разрушается. Ткань легких обычно очень тонкая и почти губчатая, потому что она заполнена воздухом в альвеолах, где кислород переходит из воздуха в кровь. Легочная ткань, пораженная бациллами туберкулеза, сначала становится твердой, что делает невозможным кислородный обмен.Это называется фиброзом.
На втором этапе клетки, составляющие легочную ткань, погибают. Это называется некрозом. Мертвая или некротическая ткань имеет тенденцию рваться и разрушаться. В легких это называется «кавитация» от латинского слова «пещера». Считается, что бациллы туберкулеза разрушают ткань легких в образовании пещеры, где все больше и больше их располагается в середине и постепенно разрушает все больше тканей по краям. Эти кавитации можно увидеть на рентгеновских снимках для диагностики туберкулеза легких (см. Рисунок).Кавитация содержит мокроту, содержащую около 1 миллиона микобактерий ТБ на миллилитр. Опасность кавитации, помимо разрушения здоровой ткани легкого, заключается в том, что в результате прогрессирующего разрушения они в конечном итоге достигают части дыхательных путей. Если это произойдет, микобактерии туберкулеза разрушают стенку дыхательных путей и, таким образом, выходят наружу. Через дыхательные пути они могут попасть в трахею, через рот и нос кашляющего человека и передаваться другим людям через воздушно-капельную инфекцию (см. Как распространяется туберкулез).
Больные туберкулезом легких часто кашляют, потому что разрушение тканей легких и дыхательных путей приводит к воспалению. Организм реагирует на воспаление, пытаясь устранить вызвавшую его частицу — если это происходит в дыхательных путях, самый простой способ устранить причину — это откашлять. Первоначально у людей с легочным туберкулезом бывает сухой постоянный кашель. Этот кашель часто усиливается ночью. Этот симптом проявляется примерно у 85% людей с легочным туберкулезом. Часто кашель сопровождается повышением температуры тела, усиливается ночью и в сочетании с чрезмерным потоотделением.Люди с туберкулезом легких теряют вес, потому что организм использует большую часть своей энергии для борьбы с инфекцией в легких, а это означает, что в организме не может храниться энергия, чтобы набрать вес и оставаться здоровым. По мере усугубления разрушения легочной ткани в мокроте, которую откашливают люди с легочным туберкулезом, появляются пятна крови — признак разрушения ткани и воспаления в дыхательных путях.
Из первой кавитации в легочной ткани бациллы ТБ могут распространяться через разрушенную ткань.Если они достигли дыхательных путей, они распространятся на другие части легких, путешествуя вверх и вниз по дыхательным путям и находя новые места для поселения и образования новой кавитации. Это называется бронхогенным распространением ( бронхов, = дыхательные пути, — генные, = исходящие). Если они получают доступ к кровеносному сосуду (кровеносные сосуды распространяются по легочной ткани для обеспечения кислородом и другими питательными веществами, транспортируемыми в крови), они могут перемещаться с кровотоком и распространяться по легким и другим частям тела. .Результат этого распространения называется «диссеминированный туберкулез» или «милиарный туберкулез», и это обычно происходит, когда иммунная система организма не в состоянии сдерживать инфекцию, например, когда она ослаблена ВИЧ.
Как туберкулез разрушает легкие | Понимание исследований на животных
28 апреля 2011
Добавил:
Категория:
Исследования и медицинские преимущества
GM мышей помогли идентифицировать ключевой фермент, ответственный за разрушение легочной ткани при туберкулезе (ТБ).Лекарства, которые ингибируют этот фермент, уже доступны, поэтому могут снизить смертность от туберкулеза.
Треть населения мира инфицирована туберкулезом, и почти 2 миллиона человек умирают от него ежегодно. Стандартное лечение оставалось неизменным в течение 35 лет, и не существует методов лечения, предотвращающих разрушение легких, вызванное туберкулезом.
ТБ вызывается бактерией M. tuberculosis . Инфекция разрушает легочную ткань пациентов, заставляя их откашливать бактерии, которые затем распространяются по воздуху и могут быть вдохнуты другими.Механизм этого повреждения легких плохо изучен. Пациентам требуется не менее шести месяцев лечения антибиотиками, но устойчивые штаммы бактерий становятся все более распространенными.
Новое исследование показывает, что у пациентов с туберкулезом повышается уровень фермента под названием MMP-1 в легких. Когда исследователи заразили туберкулезом иммунные клетки человека в лаборатории, они обнаружили, что клетки значительно увеличили выработку этого фермента.
Поскольку у мышей нет этого фермента в легких, исследователи разработали GM-мышь с человеческим MMP-1.Когда эти мыши были инфицированы туберкулезом, уровни ферментов значительно повысились, и инфекция привела к повреждению легких, аналогичному тому, что наблюдается у людей с туберкулезом. Лекарство от рака, уже доказавшее свою безопасность для людей, было эффективным в подавлении активности MMP-1, вызванной инфекцией ТБ в клетках человека.
Полученные данные позволяют предположить, что аналогичные лекарства могут предотвратить повреждение легких у больных туберкулезом и помочь ограничить распространение болезни. Планируются дальнейшие исследования.
Узнайте больше о первом лекарстве от туберкулеза.
Дата публикации
28 апреля 2011
Последний раз редактировалось: 24 сентября 2014 г. 10:36
Истощение Cd4 + Т-клеток вызывает реактивацию персистирующего туберкулеза у мышей, несмотря на продолжающуюся экспрессию интерферона и синтазы оксида азота 2
357 Scanga et al.
процессов. J. Immunol. 159: 5197–5200.
24. Роудс, E.R., A.A. Фрэнк и И.М. Орм. 1997. Прогресс-
хронический туберкулез легких у мышей аэрогенно
, инфицированных вирулентными Mycobacterium tuberculosis.Бугорок легкого
Dis. 78: 57–66.
25. Сербина, Н.В., Дж. Л. Флинн. 1999. Раннее появление
CD8⫹ Т-клеток, примированных для продукции цитокинов типа 1 в
легких мышей, инфицированных Mycobacterium tuberculosis. Заразить.
Иммун. 67: 3980–3988.
26. Бум, W.H., R.S. Уоллис, К.А. Червенак. 1991. Hu-
человек, реактивные клоны CD4⫹ Т-клеток Mycobacterium tuberculosis:
гетерогенность в распознавании антигена, продукции цитокинов,
и цитотоксичность для мононуклеарных фагоцитов.Заразить. Иммун.
59: 2737–2743.
27. Алмейда, Г.Б., К. Читале, И. Буцикакис, Ж.-Й. Geng, H.
Doo, S. He, and J.L. Ho. 1998. Индукция in vitro активности макрофагов против Mycobacterium tuberculosis человека
: потребность в интерфероне-β и примированных лимфоцитах. J. Immunol.
160: 4490–4499.
28. Лалвани А., Р. Брукс, Р. Уилкинсон, А. Малин, А. Патан,
П. Андерсен, Х. Докрелл, Г. Пасвол и А. Хилл.1998. Hu-
цитолитик человека и интерферон-гамма-секретирующий CD8⫹ T
лимфоцитов, специфичных для Mycobacterium tuberculosis. Proc. Natl.
Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 95: 270–275.
29. Левинсон Д., М. Олдерсон, А. Бриден, С. Ридделл, С. Рид,
и К. Грабштейн. 1998. Характеристика человеческих CD8⫹ T
клеток, реактивных с инфицированными Mycobacterium tuberculosis антиген-
-презентирующими клетками. J. Exp. Med. 187: 1633–1640.
30. Локсли Р.М., С.Л. Райнер, Ф. Хатам, Д. Литтман и
Н. Киллин. 1993. Хелперные Т-клетки без CD4: контроль лейшманиоза
у мышей с дефицитом CD4. Наука. 261: 1448–1451.
31. Scharton, T.M., and P. Scott. 1993. Естественные клетки-киллеры являются источником интерферона
␥, который стимулирует дифференциацию субпопуляций клеток CD4⫹ T
и индуцирует раннюю устойчивость к Leishmania major у
мышей. J. Exp. Med. 178: 567–577.
32. Gazzinelli, R.T., S.Хиени, Т.А. Wynn, S. Wolf и A.
Sher. 1993. Интерлейкин 12 необходим для независимой индукции Т-лимфоцитами-
интерферона-β внутриклеточным паразитом
и индуцирует резистентность у хозяев с дефицитом Т-клеток. Proc.
Нац. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 90: 6115–6119.
33. Fenton, M.J., M.W. Vermeulen, S. Kim, M. Burdick, R.M.
Стритер и Х. Корнфельд. 1997. Индукция продукции гамма-интер-
ферона в макрофагах человека с помощью Mycobacterium tu-
berculosis.Заразить. Иммун. 65: 5149–5156.
34. Ван Дж., Дж. Уэйкхэм, Р. Харкнесс и З. Син. 1999.
Макрофаги являются важным источником цитокинов типа 1 во время
микобактериальной инфекции. J. Clin. Вкладывать деньги. 103: 1023–1029.
35. Чан, Дж., К. Танака, Д. Кэрролл, Дж. Л. Флинн, Б. Р. Цвести.
1995. Влияние ингибиторов синтазы оксида азота на заражение мышей Mycobacterium tuberculosis. Заразить. Иммун. 63: 736–
740.
36.Ladel, C.H., S. Daugelat, and S.H.E. Кауфманн. 1995. Im-
mune ответ на инфекцию Mycobacterium bovis bacille Calmette Guerin
у мышей с основным комплексом гистосовместимости класса I- и II-
-дефицитные мыши: вклад клеток CD4 и CD8 T
в приобретенную устойчивость. Евро. J. Immunol. 25: 377–384.
37. Hansch, H.C., D.A. Смит, М.Э. Мильке, Х. Хан, Г.Дж.
Бэнкрофт и С. Элерс. 1996. Механизмы образования гранулем
при инфекции Mycobacterium avium у мышей: вклад
клеток CD4⫹ Т.Int. Иммунол. 8: 1299–1310.
38. Muller, H., and S. Kruger. 1994. Иммуногистохимический анализ
анализа клеточного состава и экспрессии цитокинов in situ при
ВИЧ- и не-ВИЧ-ассоциированных туберкулезных лимфаденитах.
Иммунобиология. 191: 354–368.
39. ДиПерри, Г., А. Каццадори, С. Венто, С. Бонора, М. Малена,
Л. Бонтемпини, М. Ланзафаме, Б. Аллегранци и Э. Конча.
1996. Сравнительное гистопатологическое исследование туберкулеза легких
беркулеза у инфицированных вирусом иммунодефицита человека и
неинфицированных пациентов.Tubercle Lung Dis. 77: 244–249.
40. Шу, У., М. Кинива, С.Ю. Wu, C. Maliszewski, N. Vezzio, J.
Kakimi, M. Gately, and G. Delespesse. 1995. Активированные клетки Т
индуцируют продукцию интерлейкина-12 моноцитами посредством взаимодействия
CD40-CD40 лиганд. Евро. J. Immunol. 25: 1125–
1128.
41. Tian, L., R.J. Ноэль, Д.А. Лоуренс. 1995. Активированные клетки T
увеличивают продукцию оксида азота макрофагами mac-
селезенки мышей через gp39 и LFI-1.Евро. J. Immunol. 25: 306–
309.
42. Кэмпбелл, К.А., П.Дж. Овендейл, М.К. Кеннеди, С.Г.
Фэнслоу, С.Г. Рид и К.Р. Малишевски. 1996. Лиганд CD40
необходим для опосредованного защитными клетками иммунитета против
Leishmania major. Иммунитет. 4: 283–289.
43. Каманака, М., П. Ю, Т. Ясуи, К. Йошида, Т. Кавабе, Т.
Хории, Т. Кишимото и Х. Кикутани. 1996. Защитная роль
CD40 в основной инфекции Leishmania на двух различных фазах
клеточно-опосредованного иммунитета.Иммунитет. 4: 275–281.
44. Soong, L., J.C. Xu, I.S. Гревал, П. Кима, Дж. Сан, Б. Дж. Лонг —
—
лей, Н. Х. Раддл, Д. МакМахон-Пратт и Р.А. Флавелл.
1996. Нарушение взаимодействий лигандов CD40-CD40 приводит к
повышенной чувствительности к инфекции Leishmania amazonensis —
. Иммунитет. 4: 263–273.
45. Кампос-Нето, А., П. Овендейл, Т. Бемент, Т.А. Коппи,
W.C. Фанслоу, М.А. Росси и М.Р. Олдерсон. 1998.
Лиганд CD40 не является существенным для развития клеточного иммунитета и устойчивости к Mycobacterium tuberculosis.J.
Immunol. 160: 2037–2041.
46. Богдан К., Ю. Водовоц и К. Натан. 1991. Макро-
дезактивация фага
интерлейкином 10. J. Exp. Med. 174: 1549–
1555.
47. Цунаваки, С., М. Спорн, А. Дин и К. Натан. 1988.
Деактивация макрофагов трансформирующим фактором роста —
бета. Природа. 334: 260–262.
48. Fauci, A.S. 1996. Факторы хозяина и патогенез заболевания, вызванного ВИЧ-
. Природа. 384: 529–534.
49. Toossi, Z., P. Gogate, H. Shiratsuchi, T. Young, and J.J. Ell-
нер. 1995. Повышенная продукция TGF-бета кровью
моноцитов от пациентов с активным туберкулезом и присутствие
TGF-бета при гранулематозных поражениях легких при туберкулезе.
J. Immunol. 154: 465–473.
50. Keene, J.A., and J. Forman. 1982. Для образования цитотоксических Т-лимфоцитов in vivo требуется хелперная активность
. J.
Exp. Med.155: 768–782.
51. von Herrath, M.G., M. Yokoyama, J. Dockter, M.B. Старый камень
и Дж. Л. Уиттон. 1996. Мыши с дефицитом CD4 имеют пониженные уровни цитотоксических Т-лимфоцитов памяти после иммунизации
и демонстрируют пониженную устойчивость к последующему заражению вирусом
. J. Virol. 70: 1072–1079.
52. Bennett, S.R., F.R. Carbone, F. Karamalis, J.F. Miller и
W.R. Heath. 1997. Для индукции ответа цитотоксических T-лимфоцитов CD8
перекрестным праймингом требуется помощь родственных клеток CD4 T
.J. Exp. Med. 186: 65–70.
53. Флинн, Дж. Л., М. М. Гольдштейн, К.Дж. Трибольд, Б. Коллер и
Б.Р. Цвести. 1992. Главный комплекс гистосовместимости, класс
I-рестриктированные Т-клетки необходимы для устойчивости к инфекции Mycobacterium
tuberculosis. Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 89: 12013–
12017.
Истощение и состав тела взрослых с туберкулезом легких в связи с коинфекцией ВИЧ-1, социально-экономический статус и тяжесть туберкулеза
Bovet P, Ross AG, Gervasoni JP, Mkamba M, Mtasiwa DM, Lengeler C et al.(2002). Распределение артериального давления, индекса массы тела и привычек курения среди городского населения Дар-эс-Салама, Танзания, и ассоциации с социально-экономическим статусом. Int J Epidemiol 31 , 240–247.
Артикул
Google ученый
Эллис К.Дж. (2000). Состав тела человека: in vivo, методов. Physiol Rev 80 , 649–680.
CAS
Статья
Google ученый
Forrester JE, Spiegelman D, Tchetgen E, Knox TA, Gorbach SL (2002).Потеря веса и изменения состава тела у мужчин и женщин, инфицированных ВИЧ. Am J Clin Nutr 76 , 1428–1434.
CAS
Статья
Google ученый
Harries AD, Nkhoma WA, Thompson PJ, Nyangulu DS, Wirima JJ (1988). Состояние питания малавийских пациентов с туберкулезом легких и ответ на химиотерапию. Eur J Clin Nutr 42 , 445–450.
CAS
PubMed
Google ученый
Карновский Д.А., Абельманн У.Х., Кравер Л.Ф., Бурченал Дж. Х. (1948).Использование азотных ипридов в паллиативном лечении рака. Рак 1 , 634–656.
Артикул
Google ученый
Кеннеди Н., Рамзи А., Уисо Л., Гутманн Дж., Нгови ФИ, Гиллеспи С.Х. (1996). Статус питания и прибавка в весе у пациентов с туберкулезом легких в Танзании. Trans R Soc Trop Med Hyg 90 , 162–166.
CAS
Статья
Google ученый
Котлер Д.П., Бурастеро С., Ван Дж., Пирсон-младший Р.Н. (1996).Прогнозирование массы клеток тела, массы без жира и общего содержания воды в организме с помощью анализа биоэлектрического импеданса: влияние расы, пола и болезни. Am J Clin Nutr 64 , 489S – 497S.
CAS
Статья
Google ученый
Котлер Д.П., Теа Д.М., Хео М., Эллисон Д.Б., Энгельсон Е.С., Ван Дж и др. (1999). Относительное влияние пола, расы, окружающей среды и ВИЧ-инфекции на состав тела взрослых. Am J Clin Nutr 69 , 432–439.
CAS
Статья
Google ученый
Котлер Д.П., Тирни А.Р., Ван Дж., Пирсон-младший Р.Н. (1989). Масштабы истощения клеточной массы тела и время смерти от истощения при СПИДе. Am J Clin Nutr 50 , 444–447.
CAS
Статья
Google ученый
Lohman TG, Roche AF, Martorell RE (1988). Справочное руководство по антропометрической стандартизации .Книги по кинетике человека: Шампейн, Иллинойс.
Google ученый
Macallan DC, McNurlan MA, Kurpad AV, de Souza G, Shetty PS, Calder AG et al. (1998). Метаболизм белков всего тела при туберкулезе легких и недостаточном питании человека: доказательства анаболической блокады при туберкулезе. Clin Sci (Лондон) 94 , 321–331.
CAS
Статья
Google ученый
Макдональд Б., Джонс Т., Грей-Дональд К., Ресивер О. (2004).Питание женщин из Анд в Эквадоре зависит от возраста и социально-экономического положения. Food Nutr Bull 25 , 239–247.
Артикул
Google ученый
Мадебо Т., Линдтьорн Б., Аукруст П., Берге РК (2003). Циркулирующие антиоксиданты и продукты перекисного окисления липидов у нелеченных больных туберкулезом в Эфиопии. Am J Clin Nutr 78 , 117–122.
CAS
Статья
Google ученый
Маллиган К., Тай VW, Шамбелан М. (1997).Поперечная и продольная оценка состава тела у мужчин с ВИЧ-инфекцией. J Acquir Syndr Immune Defic Syndr Hum Retrovirol 15 , 43–48.
CAS
Статья
Google ученый
Нийонгабо Т., Хензель Д., Иди М., Нимубона С., Гикоро Э, Мельхиор Дж. К. и др. (1999). Туберкулез, вирусная инфекция иммунодефицита человека и недоедание в Бурунди. Питание 15 , 289–293.
CAS
Статья
Google ученый
Нийонгабо Т., Млика-Кабанн Н., Барихута Т., Хензель Д., Обри П., Ларауз Б. (1994). Недоедание, туберкулез и ВИЧ-инфекция в Бурунди. СПИД 8 , 851–852.
CAS
Статья
Google ученый
Онвубалили Дж. К. (1988). Недоедание среди больных туберкулезом в Харроу, Англия. Eur J Clin Nutr 42 , 363–366.
CAS
PubMed
Google ученый
Отт М., Фишер Х., Полат Х., Хельм Е.Б., Френц М., Каспари В.Ф. и др. (1995). Анализ биоэлектрического импеданса как предиктор выживаемости у пациентов с инфекцией вируса иммунодефицита человека. J Acquir Immune Defic Syndr Hum Retrovirol 9 , 20–25.
CAS
Статья
Google ученый
Патон Н.И., Кастелло-Бранко Л.Р., Дженнингс Г., Ортигао-де-Сампайо М.Б., Элиа М., Коста С. и др.(1999). Влияние туберкулеза на состав тела ВИЧ-инфицированных мужчин в Бразилии. J Acquir Immune Defic Syndr Hum Retrovirol 20 , 265–271.
CAS
Статья
Google ученый
Патон Н.И., Чуа Ю.К., Эрнест А., Чи CB (2004). Рандомизированное контролируемое исследование пищевых добавок у пациентов с впервые выявленным туберкулезом и истощением. Am J Clin Nutr 80 , 460–465.
CAS
Статья
Google ученый
Range N, Ipuge YA, O’Brien RJ, Egwaga SM, Mfinanga SG, Chonde TM et al. (2001). Тенденция распространенности ВИЧ среди больных туберкулезом в Танзании, 1991–1998 гг. Int J Tuberc Lung Dis 5 , 405–412.
CAS
Google ученый
Скальчини М., Очченак Р., Манфреда Дж., Лонг Р. (1991). Туберкулез легких, вирус иммунодефицита человека 1 типа и недоедание. Bull Int Union Tuberc Lung Dis 66 , 37–41.
CAS
PubMed
Google ученый
Schellenberg JA, Victora CG, Mushi A, de Savigny D, Schellenberg D, Mshinda H et al. (2003). Неравенство среди очень бедных: охрана здоровья детей в сельских районах южной Танзании. Ланцет 361 , 561–566.
Артикул
Google ученый
Швенк А., Байзенхерц А., Ромер К., Кремер Г., Зальцбергер Б., Элиа М. (2000).Фазовый угол на основе анализа биоэлектрического импеданса остается независимым прогностическим маркером у ВИЧ-инфицированных пациентов в эпоху высокоактивного антиретровирусного лечения. Am J Clin Nutr 72 , 496–501.
CAS
Статья
Google ученый
Shah S, Whalen C, Kotler DP, Mayanja H, Namale A, Melikian G et al. (2001). Тяжесть инфицирования вирусом иммунодефицита человека связана со снижением фазового угла, жировой массы и массы клеток тела у взрослых с инфекцией туберкулеза легких в Уганде. J Nutr 131 , 2843–2847.
CAS
Статья
Google ученый
Suttmann U, Ockenga J, Selberg O, Hoogestraat L, Deicher H, Muller MJ (1995). Заболеваемость и прогностическое значение недоедания и истощения у амбулаторных больных, инфицированных вирусом иммунодефицита человека. J Acquir Syndr Immune Defic Syndr Hum Retrovirol 8 , 239–246.
CAS
Статья
Google ученый
Тан А.М., Форрестер Дж., Шпигельман Д., Нокс Т.А., Четген Е., Горбач С.Л. (2002).Снижение веса и выживаемость у ВИЧ-инфицированных в эпоху высокоактивной антиретровирусной терапии. J Синдр иммунодефицита Acquir 31 , 230–236.
Артикул
Google ученый
Urassa W, Matunda S, Bredberg Raden U, Mhalu F, Biberfeld G (1994). Оценка стратегии ВОЗ по тестированию антител к вирусу иммунодефицита человека (ВИЧ) для диагностики ВИЧ-инфекции. Clin Diagn Virol 2 , 1–6.
CAS
Статья
Google ученый
Ван Леттоу М., Кумвенда Дж. Дж., Харрис А.Д., Уэлен С.К., Таха Т.Э., Кумвенда Н. и др. (2004). Недоедание и тяжесть заболевания легких у взрослых с туберкулезом легких в Малави. Int J Tuberc Lung Dis 8 , 211–217.
CAS
PubMed
Google ученый
Villamor E, Msamanga G, Spiegelman D, Coley J, Hunter DJ, Peterson KE et al.(2002). ВИЧ-статус и социально-демографические корреляты размера тела матери и истощения во время беременности. Eur J Clin Nutr 56 , 415–424.
CAS
Статья
Google ученый
Белый H (1980). Оценщик ковариационной матрицы с согласованной гетероскедастичностью и прямой тест на гетероскедастичность. Econometrica 48 , 817–830.
Артикул
Google ученый
Всемирная организация здравоохранения (1998 г.). Лабораторные услуги по борьбе с туберкулезом — Часть II: Микроскопия . Всемирная организация здравоохранения: Женева.
Всемирная организация здравоохранения (2004 г.). Глобальная борьба с туберкулезом: надзор, планирование, финансирование . Отчет ВОЗ 2004: Женева.
Комитет экспертов Всемирной организации здравоохранения по физическому состоянию (1995 г.). Физический статус: использование и интерпретация антропометрии . Мировая организация здравоохранения: Женева.
Захария Р., Spielmann MP, Harries AD, Salaniponi FM (2002).Умеренное или тяжелое недоедание у больных туберкулезом является фактором риска, связанным с ранней смертью. Trans R Soc Trop Med Hyg 96 , 291–294.
CAS
Статья
Google ученый
Кислотное pH-зависимое истощение пулов тиолов Mycobacterium tuberculosis усиливает действие антибиотиков и окислителей
Резюме
Mycobacterium tuberculosis (Mtb) должно распознавать и адаптироваться к иммунному давлению, например кислому pH и реактивным формам кислорода (ROS) .Целью этого исследования было выделить соединения, которые подавляют устойчивость к кислому pH, тем самым определяя пути вирулентности, уязвимые для химиотерапии. Здесь мы сообщаем, что кислотное pH-зависимое соединение AC2P36 истощает внутриклеточные пулы тиолов, сенсибилизирует Mtb к уничтожению с помощью кислого pH и потенцирует бактерицидную активность изониазида, клофазимина и окислителей. Мы показываем, что pH-зависимая активность AC2P36 связана с метаболическим стрессом при кислом pH и pH-зависимым накоплением внутриклеточных АФК.Исследования механизма действия показывают, что AC2P36 напрямую истощает пулы тиолов Mtb. Эти данные подтверждают модель, в которой химическое истощение пулов тиолов Mtb при кислом pH повышает чувствительность к окислительному повреждению, что приводит к уничтожению бактерий и усилению действия антибиотиков.
Введение
Mycobacterium tuberculosis (Mtb), этиологический агент туберкулеза (ТБ), остается серьезной проблемой для глобального здравоохранения. Несмотря на согласованные усилия и значительные ресурсы, выделенные на борьбу с этим заболеванием, по оценкам, 10 человек.В 2015 году во всем мире произошло 4 миллиона заболеваний и 1,8 миллиона случаев смерти (ВОЗ, 2016). Основным препятствием на пути реализации стратегий борьбы с туберкулезом и его искоренения является отсутствие неизменно эффективной вакцины и появление лекарственно-устойчивого туберкулеза. Инфекции ТБ с множественной лекарственной устойчивостью являются причиной примерно 500 000 новых случаев ежегодно (ВОЗ, 2016). Возникновение лекарственной устойчивости значительно подрывает глобальный прогресс в борьбе с ТБ и подчеркивает важность разработки новых лекарств с новыми целями или механизмами действия.
Долгосрочное внутриклеточное выживание Mtb в макрофагах хозяина зависит от способности организмов адаптироваться и противостоять многочисленным стрессам окружающей среды хозяина, возникающим во время инфекции (например, окислительному, нитрозативному и кислотному стрессам). В покоящихся макрофагах Mtb ингибирует созревание фагосом и предотвращает слияние фаголизосом. Исключение вакуолярной протон-АТФазы в Mtb-содержащей вакуоли приводит к умеренно кислому фагосомному pH (pH 6,1-6,4) (MacMicking, et al., 2003; Sturgill-Koszycki, et al., 1994). Классическая активация макрофага IFN-γ устраняет эту Mtb-опосредованную блокаду, позволяя фагосомам полностью закисляться (pH 4,5-5,4) (MacMicking, et al., 2003; Schaible, et al., 1998; Via, et al., 1998). Профили экспрессии генов продемонстрировали, что в макрофагах индуцируются чувствительные к pH гены, что означает, что Mtb сталкивается с кислой средой во время инфицирования макрофагами in vitro (Fisher, et al., 2002; Rohde, et al., 2007; Rohde, et al. ., 2012). Этот кислый регулон pH очень напоминает регулон двухкомпонентной регуляторной системы (TCS) phoPR (Abramovitch, et al., 2011; Гонсало-Асенсио и др., 2008; Роде и др., 2007; Walters, et al., 2006), предполагая, что phoPR может играть роль в адаптациях, управляемых pH. Дополнительным доказательством того, что Mtb находится в подкисленной среде, является наблюдение, что пиразинамид противотуберкулезного препарата имеет повышенную активность в отношении Mtb при кислом pH (Zhang and Mitchison, 2003; Zhang, et al., 2003).
Выживание M. tuberculosis при кислом pH во время инфекции требует поддержания цитоплазматического pH-гомеостаза.Mtb может сопротивляться фаголизосомным концентрациям кислоты при выращивании в жидком бульоне, забуференном при pH 4,5-5,0, а также способен поддерживать свой цитоплазматический pH в макрофагах, активированных IFN-γ, что позволяет предположить, что Mtb обладает надежными защитными механизмами для устойчивости к кислоте (Vandal, et al. ., 2008; Чжан и др., 1999). Одной из детерминант Mtb, необходимых для кислотной устойчивости и поддержания гомеостаза pH, является ассоциированная с мембраной сериновая протеаза (Rv3671c) (Vandal, et al., 2008). Мутанты Rv3671c неспособны поддерживать нейтральный рН цитоплазмы ни в кислой ростовой среде, ни в фаголизосомах макрофагов, активированных IFN-Y (Vandal, et al., 2008). Белок, образующий поры, известный как OmpA, также важен для устойчивости к кислоте (Raynaud, et al., 2002). Экспрессия ompA индуцируется при pH 5,5, и мутанты в этом гене ослаблены для роста в макрофагах и мышах (Raynaud, et al., 2002). Наконец, предполагаемый переносчик магния (MgtC) также оказался важным для роста in vitro при умеренно кислом pH 6,25, но только в присутствии низких концентраций Mg 2+ (Buchmeier, et al., 2000). ). Нарушение MgtC привело к ослаблению роста макрофагов и мышей, что позволяет предположить, что приобретение Mg 2+ становится важным, когда Mtb подвергается воздействию низкого pH фагосомного компартмента (Buchmeier, et al., 2000).
Кислый pH вызывает широкомасштабные изменения физиологии Mtb, включая индукцию многочисленных стрессовых генов и регулона PhoPR. Кроме того, когда Mtb выращивают на одиночных источниках углерода при кислом pH, Mtb требуются источники углерода, которые питают анаплеротический узел, включая пируват, ацетат и оксалоацетат, демонстрируя, что метаболизм перестраивается при кислом pH (Baker, et al., 2014). Кислый pH также приводит к изменениям внутриклеточного окислительно-восстановительного статуса, что приводит к восстановлению среды в цитоплазме Mtb, а в отсутствие регулируемого pH TCS PhoPR цитоплазма дополнительно снижается при кислом pH, что указывает на существование pH-зависимых механизмов для поддержания окислительно-восстановительного гомеостаза.Действительно, окислительно-восстановительный датчик WhiB3 (который совместно регулирует PhoPR-регулируемые гены, такие как pks2) и окислительно-восстановительный буфер микотиол необходимы для поддержания окислительно-восстановительного гомеостаза при кислом pH и во время инфекции макрофагами (Mehta, et al., 2016; Singh, et al. др., 2009). Следовательно, выживание в условиях кислотного стресса и окислительно-восстановительный гомеостаз связаны с физиологией, хотя механизмы, управляющие pH-зависимым восстановительным стрессом, остаются плохо изученными.
Mtb подвергается воздействию кислого pH на различных стадиях во время инфекции, и эта кислая среда может действовать как критический сигнал для запуска различных адаптивных механизмов, необходимых для выживания в организме хозяина.Таким образом, цель этого исследования состояла в том, чтобы идентифицировать и охарактеризовать соединения, которые избирательно ингибируют рост Mtb при кислом pH. Эти соединения могут идентифицировать новые лекарственные мишени или пути, которые необходимы для роста при кислом pH, которые были бы упущены при ранее описанных проверках лекарств, проводимых в богатой среде при нейтральном pH. Наша команда провела два независимых высокопроизводительных скрининга для поиска ингибиторов кислотных pH-индуцируемых или индуцируемых гипоксией сигнальных путей (Johnson, et al., 2015; Чжэн и др., 2016). Скрининг проводился в почти идентичных условиях, включая одну и ту же библиотеку ~ 220 000 соединений, с основным различием в pH среды (pH 7,0 или pH 5,7). Сравнение попаданий между двумя экранами выявило несколько соединений, которые подавляют рост Mtb при кислом pH, но не влияют на нейтральный pH. Мы предположили, что pH-зависимые соединения могут быть нацелены на пути, которые необходимы для роста только при кислом pH. Здесь мы сообщаем, что одно из pH-зависимых соединений, идентифицированных при скрининге, названное AC2P36, убивает Mtb при кислом pH, истощает внутриклеточные пулы тиолов, способствует накоплению внутриклеточных ROS и потенцирует активность изониазида, клофазимина и окислителей.Эти данные подтверждают, что пути окислительно-восстановительного гомеостаза являются уязвимыми pH-зависимыми мишенями и при их ингибировании способствуют уничтожению Mtb и усилению действия противомикробных препаратов.
Результаты
Открытие AC2P36 в качестве кислого pH-зависимого ингибитора роста Mtb
Два полноценных фенотипических высокопроизводительных скрининга (HTS) были проведены при нейтральном и кислом pH для определения ингибиторов регулонов DosRST и PhoPR, соответственно, с использованием идентичная химическая библиотека (Johnson, et al., 2015; Zheng, et al., 2016). Библиотека скрининга ~ 220 000 соединений состоит из небольших молекул, представляющих широкое химическое разнообразие. Изучая перекрытие между ингибиторами, которые снижают рост Mtb, были обнаружены соединения, которые избирательно ингибируют рост Mtb при кислом pH. Небольшие молекулы, которые демонстрировали профиль снижения роста по меньшей мере на 50% и были уникальными для HTS при кислом pH, рассматривались для дополнительного наблюдения и характеризации как pH-зависимые ингибиторы роста Mtb. AC2P36 (5-хлор-N- (3-хлор-4-метоксифенил) -2-метилсульфонилпиримидин-4-карбоксамид, рис.1A) был идентифицирован как соединение, которое соответствовало профилю замедления роста Mtb при кислом pH, но не влияло на рост при нейтральном pH. При pH 5,7 AC2P36 ингибирует рост Mtb с полумаксимальной эффективной концентрацией (EC 50 ) ~ 3 мкМ, тогда как при pH 7,0 EC 50 составляет> 30 мкМ (рис. 1B), таким образом, AC2P36 показывает ~ 10 -кратная селективность при кислом pH. При концентрациях> 30 мкМ рост Mtb замедляется при нейтральном pH, что позволяет предположить, что механизм действия AC2P36 не может быть исключительным для кислого pH.AC2P36 не модулирует pH цитоплазмы Mtb, что указывает на то, что он не функционирует как ионофор (дополнительный рисунок 1A). Чтобы оценить, действует ли AC2P36 бактерицидным или бактериостатическим образом при кислом pH, были предприняты зависимые от времени и дозозависимые анализы выживаемости. AC2P36 в концентрации 20 мкМ демонстрирует зависящее от времени уничтожение, вызывая прогрессивное ~ 100-кратное снижение жизнеспособности в течение 7 дней (рис. 1C). Бактерицидная активность AC2P36 зависит от дозы, при этом бактерицидная активность наблюдается при концентрациях> 10 мкМ после 6 дней лечения (рис.1D). Mtb демонстрирует восстановленную цитоплазму при кислом pH и требует PhoPR для поддержания окислительно-восстановительного баланса (Baker, et al., 2014). Чтобы проверить, является ли способность AC2P36 убивать Mtb при кислом pH результатом измененного окислительно-восстановительного гомеостаза, Mtb, экспрессирующий окислительно-восстановительный активный GFP, обрабатывали возрастающими концентрациями AC2P36. Обработка AC2P36 приводит к уменьшению цитоплазмы дозозависимым и pH-независимым образом (рис. 1E). Поскольку было показано, что PhoPR необходим для pH-зависимого поддержания окислительно-восстановительного гомеостаза, мы предположили, что мутант ∆phoPR может быть более восприимчивым к AC2P36, однако никаких различий между чувствительностью WT или ∆phoPR к AC2P36 при кислом pH не наблюдалось. (Рис.1F). Эти данные демонстрируют, что AC2P36 ингибирует рост Mtb pH-зависимым образом, обладает бактерицидным действием и способствует уменьшению цитоплазмы.
Рис. 1. AC2P36 представляет собой pH-селективное бактерицидное соединение, которое способствует уменьшению внутриклеточной среды.
A) Химическая структура AC2P36 (5-хлор-N- (3-хлор-4-метоксифенил) -2-метилсульфонилпиримидин-4-карбоксамид). B) Обработка AC2P36 ингибирует рост Mtb дозозависимым образом при pH 5,7 с EC 50 3,2 мкМ. Рост бактерий подавляется при pH 7.0 дозозависимым образом при концентрациях более 30 мкМ. C) Зависящее от времени уничтожение Mtb при pH 5,7, обработанных 20 мкМ AC2P36. D) Дозозависимое уничтожение Mtb при pH 5,7 после 6 дней лечения AC2P36. Пунктирной линией обозначены КОЕ исходного посевного материала. E) AC2P36 способствует уменьшению цитоплазмы дозозависимым и pH-независимым образом. Цитоплазматический окислительно-восстановительный статус измеряли с помощью редокс-чувствительного ro-GFP. F). CDC1551 (AphoPR) имеет такую же чувствительность к AC2P36, как и WT CDC1551.Показанные данные являются репрезентативными, по крайней мере, для двух независимых экспериментов, а полосы ошибок представляют собой стандартное отклонение среднего значения.
AC2P36 проявляет узкий спектр активности
Чтобы оценить, действует ли AC2P36 как широкий ингибитор роста бактерий при кислом pH, выбранные грамположительные и грамотрицательные бактерии обрабатывались соединением при нейтральном и кислом pH (дополнительная таблица 1 ). Только грамположительные виды Staphylococcus aureus и Enterococcus faecalis проявляют восприимчивость к AC2P36 (дополнительная таблица 1), тогда как грамотрицательные бактерии видов Escherichia coli, Pseudomonas auruginosa, и 9000a0006 Proteus были относительно незараженными.Примечательно, что как у S. aureus, так и у E. faecalis чувствительность AC2P36 не зависела от pH, и соединение было менее активным. Вместе эти данные предполагают, что Mtb избирательно чувствителен к AC2P36, но что общий путь может быть нацелен на Mtb, S. aureus и E. faecalis .
AC2P36 вызывает тиол-ассоциированный стрессовый ответ
Чтобы дополнительно охарактеризовать механизм действия AC2P36, было выполнено глобальное профилирование транскрипции для выявления путей, которые дифференциально экспрессируются в ответ на лечение AC2P36.Mtb инокулировали в богатую среду, забуференную при pH 7,0 или 5,7, в присутствии либо 10 мкМ AC2P36, либо эквивалентного объема ДМСО. После 4 часов инкубации общую РНК выделяли и подвергали РНК секвенированию. Обработка Mtb AC2P36 вызвала индукцию 167 генов (> 2-кратное, q <0,05) и 180 генов (> 2-кратное, q <0,05) при pH 7,0 и pH 5,7 (рис. 2A, дополнительные таблицы S2-S4. ), соответственно. Примечательно, что 124 гена были индуцированы как при pH 7,0, так и при pH 5,7, что указывает на то, что AC2P36 модулирует физиологию Mtb независимо от pH (рис.2Б). Многие из генов, активируемых при любом pH, также регулируются альтернативным сигма-фактором sigH в ответ на окислительный / тиоловый стресс или перекисью водорода (рис. 2ABC) (Manganelli, et al., 2002; Voskuil, et al. , 2011). Действительно, sigH был значительно активирован (> 5 раз, q <0,05), включая несколько генов в его регулоне, таких как тиоредоксинредуктазы ( trxB, trxB2, trxC ), гены синтеза липидов ( papA4 ), цистеин синтез ( cysM ) и несколько предсказанных оксидоредуктаз ( Rv2454c , Rv3463 ).AC2P36 также в значительной степени индуцирует дополнительные гены, связанные с физиологией, связанной с сульфуром, включая транспорт сульфата ( cysA , cysW , cysT ) и тиосульфат-сертрансферазу ( sseC2 ). Эти данные убедительно указывают на то, что AC2P36 может действовать, направляя действие на гомеостаз тиолов. Примечательно, что транскрипционный ответ окислительного стресса, связанный с детоксикацией супероксидных видов посредством разобщающего комплекса супероксиддисмутазы ( sodA , sodC ) или мембранно-ассоциированной оксидоредуктазы ( sodA , sseA , doxX ), не наблюдался, общий ответ на окислительный стресс от лечения AC2P36 (Nambi, et al., 2015). Однако каталаза / пероксидаза katG и регулятор захвата железа furA активируются более чем в 4 раза при pH 7,0 и 5,7, а многочисленные регулируемые гены H 2 O 2 индуцируются AC2P36, что позволяет предположить, что реактивный кислород виды могут образовываться и требовать детоксикации. Взятые вместе, эти данные предполагают, что AC2P36 влияет на транскриптом Mtb как при нейтральном, так и при кислом pH. Однако повышенная чувствительность Mtb к обработке AC2P36 при кислом pH, вероятно, связана с pH-зависимой физиологией, возможно, через ранее описанную pH-зависимую регуляцию гомеостаза тиолов Mtb (Baker, et al., 2014; Бухмайер и др., 2006; Mehta, et al., 2016).
Рисунок 2. AC2P36 стимулирует гены, связанные с регулятором sigH и окислительным стрессом.
A) График величины-амплитуды RNA-seq для Mtb, обработанного 10 мкМ AC2P36 в течение 4 часов (pH 5,7). Выделенные гены участвуют в альтернативном сигма-факторе , регулоне sigH , а katG представляет собой каталазу / пероксидазу Mtb. Черные точки не являются статистически значимыми, а красные точки статистически значимыми (q <0.05). B) Диаграмма Венна для генов, активируемых (> 2 раза, q <0,05) в Mtb, обработанном AC2P36, при pH 7,0 и 5,7, перекрывающихся с регулятором sigH или регулируемыми генами H 2 O 2 (Manganelli, et al. , 2002; Воскуил и др., 2011). Обратите внимание на значительное совпадение как нейтрального, так и кислого pH в культурах, обработанных AC2P36. C) Тепловая карта, показывающая гены регулона sigH, которые индуцируются (> 2 раза, q <0,05) AC2P36. Данные представляют собой среднее значение двух биологических повторов. Сравнение проводится только с генами, аннотированными в геноме h47Rv.
Данные транскрипционного профилирования предполагают, что AC2P36 способствует тиол-ассоциированной стрессовой реакции, поэтому мы предположили, что AC2P36 может нарушать окислительно-восстановительный баланс бактерий и повышать чувствительность Mtb к окислителям и антимикробным препаратам, таким как диамид, клофазимин и изониазид (INH). Чтобы определить, вызывает ли AC2P36 повышенную чувствительность к окислению тиола диамидом, Mtb инокулировали в богатую среду, забуференную при pH 5,7, в присутствии или в отсутствие 10 мкМ AC2P36 в комбинации либо с 1 мМ диамидом, либо с эквивалентным объемом ДМСО.После 3 дней обработки культуры высевали на чашки с агаром 7h20, и жизнеспособность бактерий оценивали путем подсчета колониеобразующих единиц (КОЕ). Обработка только AC2P36 или диамидом приводила к приблизительно 2-кратному снижению или 1,8-кратному усилению роста бактерий по сравнению с исходным посевным материалом, соответственно (фиг. 3A). Однако комбинация диамида и AC2P36 приводила к более чем 8-кратному увеличению киллинга по сравнению с исходным посевным материалом, демонстрируя потенцирующие взаимодействия соединений (рис.3А). Это открытие подтверждает, что AC2P36 может повышать чувствительность Mtb к окислению тиолов. Клофазимин, компонент схемы комбинированного лечения инфекции Mycobacterium leprae , представляет собой соединение с окислительно-восстановительным циклом, которое может напрямую конкурировать с менахиноном и спонтанно продуцировать реактивные формы кислорода (Lechartier and Cole, 2015). Если функция AC2P36 истощает пулы тиолов и снижает устойчивость к окислительному стрессу, то предполагается, что AC2P36 повышает чувствительность Mtb к клофазимину. Чтобы проверить эту гипотезу, 10 мкМ AC2P36 инкубировали в сочетании со 100 мкМ клофазимином в течение 3 дней при pH 5.7. Лечение клофазимином само по себе не влияло на рост Mtb, однако клофазимин в комбинации с AC2P36 приводил примерно к 22-кратному снижению роста бактерий (фиг. 3A). Эти данные предполагают, что лечение AC2P36 нарушает механизмы устойчивости к окислителям. Кроме того, учитывая индукцию katG с помощью AC2P36, мы предположили, что AC2P36 может сенсибилизировать Mtb к INH (пролекарство, которое активируется KatG). Чтобы проверить эту гипотезу, Mtb обрабатывали в течение 3 дней при высоком pH 5.7 в буферной среде в присутствии или в отсутствие 10 мкМ AC2P36 в комбинации с 0,3 мкМ рифампицином, 10 мкМ INH или эквивалентным объемом ДМСО. Обработка AC2P36 и только INH приводила к сравнимым уровням уничтожения с приблизительно 2-кратным снижением роста бактерий по сравнению с исходным посевным материалом (фиг. 3A). Однако в комбинации AC2P36 и INH действуют синергично, снижая выживаемость бактерий примерно в 100 раз (рис. 3A). Лечение AC2P36 не имело значительного синергизма с другим противотуберкулезным препаратом, рифампицином, в тестируемой концентрации (рис.3А). Вместе эти данные показывают, что AC2P36 усиливает бактерицидную активность окислителей и специфических антибиотиков.
Рис. 3. AC2P36 сенсибилизирует Mtb к обработке окислителями, способствует накоплению внутриклеточных АФК и истощает свободные тиолы.
A) Обработка Mtb 10 мкМ AC2P36 в сочетании с диамидом, окисляющим тиол, снижает рост примерно в 8 раз по сравнению с исходным посевным материалом. Кроме того, агент окислительно-восстановительного цикла клофазимин также усиливает лечение AC2P36, что приводит к ~ 24-кратному снижению роста Mtb.Комбинированное лечение AC2P36 и INH приводит к усилению уничтожения бактерий (~ 160-кратное снижение роста бактерий). В комбинации с антибиотиком рифампицином никаких эффектов не наблюдалось. Данные являются репрезентативными для двух биологических повторов, и все различия являются статистически значимыми (t-тесты, множественные сравнения с поправкой, p <0,05), если не указано иное. н.с. - статистически не значимо. B) Обработка Mtb AC2P36 приводит к дозозависимому и pH-зависимому накоплению активных форм кислорода (ROS).ROS измеряли с использованием флуоресцентного красителя CellROX Green (5 мкМ) и нормализовали по оптической плотности (OD 595 ). * представляет статистическую значимость (p <0,05, 2-сторонний дисперсионный анализ). C) Mtb, обработанный указанными концентрациями AC2P36, приводит к истощению пулов тиолов по сравнению с контролем ДМСО. Ауранофин - известный ингибитор, который снижает концентрацию свободных тиолов, в то время как изониазид (INH) - противотуберкулезный антибиотик, который напрямую не истощает свободные тиолы. Статистическая значимость рассчитывалась на основе двустороннего t-критерия (*, p <0.05). Для каждой панели планки ошибок представляют стандартное отклонение среднего значения.
AC2P36 вызывает накопление внутриклеточных активных форм кислорода
Данные транскрипционного профилирования и потенцирования окислительных агентов предполагают, что AC2P36 может способствовать накоплению внутриклеточных активных форм кислорода (ROS). Недавние исследования показали, что увеличение эндогенных АФК может иметь значительные и пагубные последствия для роста и жизнеспособности Mtb in vitro и во время инфекции (Vilcheze, et al., 2013) (Тяги и др., 2015). Для измерения образования внутриклеточных АФК в Mtb использовали флуоресцентный краситель CellROX green (Saini, et al., 2016). Mtb выращивали в богатой среде, осаждали и ресуспендировали в буферной среде 7H9 с pH 7,0 или 5,7. Культуры инкубировали в присутствии 2 или 20 мкМ AC2P36 или эквивалентного объема ДМСО. Как при кислом, так и при нейтральном pH AC2P36 вызывал значительную индукцию ROS (рис. 3B). AC2P36 в сочетании с кислым pH удваивает накопление внутриклеточных ROS по сравнению с клетками, обработанными AC2P36 при нейтральном pH.Примечательно, что один только кислый pH вызывает накопление ROS, предполагая, что стресс, связанный с кислым pH, вызывает усиленное образование ROS или ингибирует механизмы устойчивости к окислительному стрессу. Чтобы проверить гипотезу о том, что кислый pH снижает способность Mtb противодействовать окислительному стрессу, мы исследовали внутриклеточное накопление ROS в обработанных диамидом клетках при кислом pH. Диамид в концентрации 20 мкМ вызывает значительное pH-зависимое увеличение ROS при pH 5,7. Эти данные согласуются с тем, что Mtb имеет пониженную устойчивость к окислительному стрессу при кислом pH.
AC2P36 истощает внутриклеточные пулы тиолов
Мы выдвинули гипотезу, что AC2P36 может истощать пулы тиолов кислым pH-зависимым образом, учитывая, что AC2P36 способствует тиоловому стрессу и чувствительности к окисляющим агентам, и что кислотный pH, как было показано, способствует восстановительному стрессу и увеличивает накопление низкомолекулярного тиолового буфера микотиола (Buchmeier, et al., 2006). Для прямого измерения пулов свободных тиолов внутри клетки Mtb инокулировали в богатую среду, забуференную при pH 7.0 и pH 5,7 в присутствии 2 мкМ AC2P36, 20 мкМ AC2P36, 20 мкМ ауранофина, INH или эквивалентного объема ДМСО. После 24-часовой инкубации культуры анализировали на общие пулы внутриклеточных тиолов. Предыдущая работа показала, что ауранофин истощает свободные тиолы в Mtb и S. aureus; , поэтому его использовали в качестве положительного контроля (Harbut, et al., 2015). INH использовался в качестве отрицательного контроля, поскольку не ожидается, что он напрямую истощит свободные тиолы. Действительно, свободные тиолы с истощением ауранофина по сравнению с контролем, обработанным ДМСО, и INH имели сравнимые концентрации тиолов с необработанными культурами (рис.3С). AC2P36 значительно истощил пулы тиолов при pH 7,0 и 5,7. При pH 5,7 20 мкМ AC2P36 истощали свободные тиолы от ~ 16 раз до ~ 80 нМ по сравнению с ~ 1,3 мкМ в образцах, обработанных ДМСО (рис. 3C), тогда как при pH 7,0 свободные тиолы уменьшались только на одну треть. В целом эти данные показывают, что AC2P36: i) способствует сокращению цитоплазмы, ii) увеличивает внутриклеточные ROS и iii) значительно истощает пулы свободных тиолов с усиленным истощением при кислом pH. Эти данные демонстрируют, что Mtb более чувствителен к тиоловым агентам при кислом pH, и что эта pH-зависимая чувствительность увеличивает уязвимость к окислителям и антибиотикам.Полученные данные согласуются с предыдущими сообщениями о том, что мутанты Mtb или M. smegmatis , дефицитные по микотиолу, очень чувствительны к агентам окислительно-восстановительного цикла, H 2 O 2 или аскорбиновой кислоте (витамин C) (Buchmeier, et al., 2006; Rawat, et al., 2002; Vilcheze, et al., 2013). Интересно, что было показано, что мутанты микотиола обладают повышенной устойчивостью к INH (Vilcheze, et al., 2011; Xu, et al., 2011) (Rawat, et al., 2002), в отличие от потенцирования INH, вызванного AC2P36, Это указывает на то, что механизм действия AC2P36 не влияет исключительно на путь окислительно-восстановительного цикла микотиола.
AC2P36 модулирует физиологию
S. aureus с помощью механизмов, аналогичных Mtb
. Учитывая, что AC2P36 может также ингибировать рост S. aureus , мы исследовали, увеличивает ли AC2P36 внутриклеточные АФК и истощает свободные тиолы S. aureus. Для исследования внутриклеточных ROS S. aureus инокулировали в богатую среду, забуференную до pH 7,0 или 5,7, и подвергали серии 4-точечных (2-кратных) разведений AC2P36, DTT и менадиона в диапазоне от 10 до 80 мкМ или эквивалентный объем ДМСО.После 6 часов инкубации культуры оценивали на накопленные ROS. AC2P36 вызывал повышенное накопление ROS дозозависимым образом при pH 7,0 и 5,7, аналогично положительному контролю с менадионом (фиг. 4A). Примечательно, что ROS накапливаются на более высоких уровнях при нейтральном pH по сравнению с кислым pH, что указывает на различия в pH-зависимом окислительно-восстановительном гомеостазе между Mtb и S. aureus. AC2P36 также значительно снижает количество свободных тиолов в S. aureus, с 20 мкМ AC2P36 или ауранофином, что приводит к снижению количества свободных тиолов примерно на 30% (рис.4Б). Повышенное накопление ROS может непосредственно приводить к торможению роста. Чтобы проверить эту гипотезу, чувствительность к AC2P36 была протестирована с мутантом S. aureus ( ΔsodAΔsodM ), дефицитным по супероксиддисмутазе, который имеет пониженную устойчивость к окислительному стрессу (Kehl-Fie, et al., 2011). По сравнению с диким животным, S. aureus ( ΔsodAΔsodM ) продемонстрировал примерно в 2 раза повышенную чувствительность к AC2P36 при рН 7,0 и 5,7 (рис. 4С). Эти данные предполагают, что обработка AC2P36 приводит к накоплению внутриклеточных АФК в S.aureus , и это ингибирование роста частично обусловлено механизмом, опосредованным ROS.
Рис. 4. AC2P36 стимулирует продукцию ROS и истощает свободные тиолы в S. aureus .
A) Обработка S. aureus указанными концентрациями AC2P36 приводит к дозозависимому накоплению активных форм кислорода (ROS). В отличие от Mtb, S. aureus демонстрирует повышенное накопление ROS при нейтральном pH. S. aureus , обработанный AC2P36, демонстрирует аналогичную дозозависимую реакцию на агент менадиона, вызывающий окислительно-восстановительный цикл.ДТТ не влиял на накопление АФК. ROS измеряли с использованием флуоресцентного красителя CellROX Green (5 мкМ) и нормализовали по оптической плотности (OD 595 ). Все различия в S. aureus являются статистически значимыми между контрольными клетками, обработанными ДМСО, и клетками, обработанными AC2P36 (p <0,05, двухфакторный дисперсионный анализ). B) S.aureus , обработанный указанными концентрациями AC2P36 при pH 5,7, приводит к истощению пулов тиолов по сравнению с контролем ДМСО. Статистическая значимость рассчитывалась на основе двустороннего t-критерия (*, p <0.05). C) Чувствительность S. aureus к AC2P36 повышена примерно в 2 раза у мутанта S. aureus (AsodAAsodM) как при pH 7,0, так и при pH 5,7, подтверждая, что ингибирование роста частично вызвано окислительным стрессом.
Сульфоновая группа AC2P36 необходима для активности
Эукариотическая цитотоксичность AC2P36 была протестирована на макрофагах J774, и было обнаружено, что соединение имело полумаксимальную концентрацию цитотоксичности (CC 50 ) 17 мкМ. Поэтому мы стремились определить химические группы, необходимые для активности против Mtb, минимизируя цитотоксические эффекты против клеток млекопитающих.Чтобы определить реактивные части AC2P36, была определена химическая структура ядра и несколько аналогов исходной молекулы (AC2P36-A, фиг. 5) были оценены на бактерицидную активность или эукариотическую цитотоксичность (фиг. 5). Изменения химических групп в областях R1 и R2 структуры ядра показали, что сульфоновый фрагмент в положении R1 необходим для активности против Mtb при pH 5,7. В аналогах AC2P36-H и AC2P36-I метилсульфоновая группа заменена альтернативными группами, что делает обе молекулы неактивными по отношению к Mtb.Примечательно, что аналог AC2P36-J показывает, что фрагмент R2 не требуется для уничтожения Mtb. Кроме того, этот аналог AC2P36-J не проявляет цитотоксичности в отношении макрофагов вплоть до самой высокой испытанной концентрации (80 мкМ), что позволяет предположить, что группа 5-хлор-N- (3-хлор-4-метоксифенил) R2 может управлять цитотоксичностью исходное соединение; хотя нельзя исключить, что потеря фрагмента R2 может ограничивать проникновение аналога AC2P36-J в макрофаг. Эти данные предполагают, что реакционноспособная группа AC2P36 находится в метилсульфонилпиримидиновом каркасе и что сульфоновый фрагмент необходим для активности.
Рисунок 5. Исследования взаимосвязи структуры и активности AC2P36.
Тестирование ингибирования роста и цитотоксичности макрофагов (CC 50 ) исходного аналога AC2P36-A и 9 дополнительных аналогов (B-J) с вариациями в указанных группах R1 и R2. Группа R2 необязательна, в то время как группа R1 необходима для ингибирования роста Mtb при pH 5,7. Примечательно, что эукариотическая цитотоксичность устранена в активном аналоге Mtb AC2P36-J.
AC2P36 напрямую нацелен на тиолы
Основной химический каркас AC2P36 (5-хлор-2-метилсульфонилпиримидин-4-карбоксамид) был ранее исследован для синтеза производных каркасов аминопиримидинсодержащих лекарственных средств (Moon, et al., 2013). Примечательно, что анионный тиолат нуклеофила селективно реагирует с углеродом C-2 почти исключительно в зависимости от электроноакцепторной группы (Moon, et al., 2013). В случае метилсульфонового фрагмента эта химическая группа является сильной электроноакцепторной группой, способствующей частичному положительному заряду на углероде C-2, позволяя богатому электронами тиолат-аниону атаковать и замещать метилсульфон (Bebbington, et al., 2009 ). Основываясь на известной химии, мы предположили, что AC2P36 может напрямую воздействовать на тиолы.Чтобы проверить эту гипотезу, мы инкубировали 80 мкМ AC2P36 со 100 мкМ восстановленным глутатионом (GSH) или контролем ДМСО как в кислых, так и в нейтральных условиях в течение одного часа. Затем образцы исследовали масс-спектрометрией. В образцах, обработанных только AC2P36, AC2P36 был обнаружен как пик с массой 373,976 Да. Однако в образцах, обработанных AC2P36 и GSH как при кислотном, так и при нейтральном pH, новый пик наблюдался при 601,066 Да (фиг. 6A). Этот новый пик согласуется с предложенной реакцией, показанной на Фигуре 6B, где ковалентная модификация свободных тиолов AC2P36 способствует высвобождению либо метансульфиновой кислоты, либо метансульфината.Например, объединенные массы AC2P36 (373,976 Да) и GSH (307,32 Да) составляют 681,246 Да, а потеря метансульфиновой кислоты (79,065 Да) и протона приводит к молекуле с массой 601 Да. Эти данные подтверждают, что AC2P36 способен образовывать стабильные аддукты со свободными тиолами, и подтверждают предложенный механизм реакции.
Рис. 6. AC2P36 образует ковалентные аддукты со свободными тиолами.
A) Инкубация AC2P36 с восстановленным глутатионом (GSH) приводит к образованию аддуктов AC2P36-GSH, о чем свидетельствует образование новой молекулы с молекулярной массой ~ 601 Да.Б) Предлагаемый механизм реакции, который может генерировать наблюдаемый аддукт AC2P36-GSH. НАДФН-зависимая активация общего дисульфида в тиол и тиолат-анион позволяет тиолату подвергаться анионной нуклеофильной атаке углерода C-2 на пиримидиновое кольцо AC2P36, что приводит к ковалентной модификации и высвобождению либо метансульфиновой кислоты, либо метансульфинита.
Обсуждение
AC2P36 был обнаружен как pH-зависимый ингибитор роста Mtb путем проведения двух цельноклеточных HTS из идентичной библиотеки соединений при нейтральном и кислом pH.Исследования механизма действия подтверждают, что AC2P36 истощает внутриклеточные пулы тиолов и способствует внутриклеточному накоплению ROS, и эта активность может привести к наблюдаемой бактерицидной активности против Mtb. Примечательно, что Mtb демонстрирует уменьшенные пулы тиолов, накопление ROS и связанные с ними изменения в экспрессии генов как при кислом, так и при нейтральном pH, подтверждая, что AC2P36 модулирует физиологию Mtb независимо от pH. Однако величина возмущений увеличивается при кислом pH, и Mtb селективно уничтожается AC2P36 при кислом pH.Таким образом, мы предполагаем, что Mtb обладает повышенной чувствительностью к тиоловому стрессу, вызванному AC2P36, при кислом pH. Эти наблюдения подтверждаются недавними исследованиями, показывающими, что кислый pH способствует метаболическому стрессу, связанному с уменьшением цитоплазмы (Baker, et al., 2014; Mehta, et al., 2016). Предыдущее исследование показало, что пулы микотиолов увеличиваются примерно в два раза при кислом pH (pH 5,5), предполагая, что Mtb адаптируется к тиоловой нагрузке при кислом pH (Buchmeier, et al., 2006). Примечательно, что анализы пула тиолов, проведенные в этом исследовании (рис.3C) не обнаруживали изменений в пулах тиолов при кислотном pH по сравнению с нейтральным в контрольных образцах, обработанных ДМСО. Однако между исследованиями существует несколько различий, в том числе продолжительность кислотного стресса и чувствительность тестов для обнаружения тиолов (Buchmeier, et al., 2006).
Учитывая, что Mtb испытывает уменьшение цитоплазмы при кислом pH, мы предлагаем модель, в которой повышенная чувствительность Mtb к AC2P36 при pH 5,7 является результатом большей доли пулов тиолов, находящихся в восстановленном состоянии. Затем AC2P36 может ковалентно модифицировать восстановленные тиолы, тем самым подавляя их способность к повторному окислению (рис.7). Этот модельный механизм подтверждается данными GFP, чувствительными к окислительно-восстановительному потенциалу, указывающими на уменьшение цитоплазмы после обработки AC2P36 (рис. 1E), результатами профилирования транскрипции, указывающими на усиленный восстановительный стресс, опосредованный тиолами и pH-зависимыми AC2P36, и большее истощение пулов тиолов в кислой среде. pH относительно нейтрального pH (рис. 3C). Учитывая, что мы не наблюдали повышенных уровней свободных тиолов при кислом pH, возможно, что уменьшенная внутриклеточная среда достаточна для поддержания пулов тиолов в уменьшенном и, следовательно, в чувствительном к AC2P36 состоянии.Пути рециркуляции тиолов играют важную роль в противодействии окислительному стрессу, поэтому следует, что AC2P36 может ингибировать способность Mtb рассеивать окислительный стресс и, таким образом, приводить к наблюдаемому усиленному накоплению ROS и снижению устойчивости к окислителям. Мы предполагаем, что нечувствительность Mtb к AC2P36 при нейтральном pH обусловлена более окисленной цитоплазмой, что приводит к появлению AC2P36-устойчивых окисленных тиолов, или, альтернативно, бактериям может быть доступна дополнительная устойчивость к АФК или пути рециркуляции тиолов при нейтральном pH.Предполагается, что Mtb займет кислые ниши во время инфекции, поэтому наши результаты являются доказательством концептуального исследования, показывающего, что нацеливание на свободные пулы тиолов является потенциальной терапевтической стратегией для разработки лекарств Mtb.
Рис. 7. Модельный механизм AC2P36-зависимого ингибирования роста и потенцирования окислителей.
A) Нормальные ответы Mtb на стресс реактивных форм кислорода (ROS) при нейтральном и кислом pH могут происходить за счет рециркуляции пулов окисленных тиолов через низкомолекулярный тиол (RSH) и микоредоксин / тиоредоксин (не показан) для регенерации дисульфидной связи .B) При нейтральном pH AC2P36 истощает пулы тиолов, но Mtb обладает способностью поддерживать или регенерировать пулы тиолов для защиты от ROS. Однако при кислом pH Mtb имеет восстановленную цитоплазму, что приводит к повышенному отношению восстановленных тиолов к окисленным дисульфидам. Обработка AC2P36 ковалентно модифицирует свободные тиолы, не позволяя Mtb поддерживать или восстанавливать внутриклеточные пулы тиолов и окислительно-восстановительную буферизацию. В результате Mtb неспособен защищаться от окислительного повреждения, что приводит к гибели клеток и усилению действия окислителей.
AC2P36 сильно индуцирует многие гены в альтернативном сигма-факторе SigH-регулоне, указывая на то, что Mtb испытывает тиоловый стресс при обработке AC2P36 (Manganelli, et al., 2002). Реглон SigH был идентифицирован на основании сравнения диамида окислителя тиола с обработанным диамидом мутантом sigH . Индуцированные гены в регулоне SigH включают тиоредоксины, тиоредоксинредуктазу, а также гены, связанные с биосинтезом цистеина. Тиоредоксины и тиоредоксинредуктаза являются ключевыми ферментами в процессе поддержания окислительно-восстановительного гомеостаза в Mtb.Если способность рециркулировать тиолы через тиоредоксинредуктазу ингибируется, Mtb ослабляется для роста (Harbut, et al., 2015). Однако AC2P36, вероятно, не ингибирует напрямую тиоредоксинредуктазу, поскольку этот фермент важен для роста независимо от pH (Harbut, et al., 2015). Наблюдаемая повышающая регуляция тиоредоксинов и тиоредоксинредуктазы при pH 7,0 и 5,7 предполагает, что AC2P36 потенциально блокирует способность Mtb рециркулировать свой пул тиолов для поддержания окислительно-восстановительного баланса. Кроме того, усиление биосинтеза цистеина вовлечено в поддержание окислительно-восстановительного баланса в штаммах Mtb, дефицитных по низкомолекулярному тиолэрготионеину и окислительно-восстановительному сенсору WhiB3 (Saini, et al., 2016). Однако мы не наблюдали повышения регуляции оперонов биосинтеза микотиола или эрготионеина под действием AC2P36 ни при нейтральном, ни при кислом pH, что позволяет предположить, что активность AC2P36 напрямую не способствует увеличению продукции низкомолекулярных тиолов в качестве контрмеры. Повышение регуляции многофункциональной каталазы / пероксидазы KatG может частично объясняться окислением NADH с помощью KatG для поддержания окислительно-восстановительного баланса и противодействия активности AC2P36 (Singh, et al., 2004). Однако эта функция KatG также потенциально вредна для клетки, поскольку перекись водорода вырабатывается в процессе потребления восстанавливающих эквивалентов (Farhana, et al., 2010; Сингх и др., 2004). Мы наблюдаем как повышенное накопление ROS, так и ~ 100-кратное усиление INH, когда Mtb обрабатывают AC2P36, и, таким образом, предполагаем, что индукция KatG может управлять повышенной чувствительностью к пролекарству INH, хотя дополнительные механизмы, вероятно, управляют синергетическим взаимодействием. Примечательно, что ранее было показано, что модуляция гомеостаза тиолов путем манипулирования синтезом микотиола вызывает устойчивость к INH (Rawat, et al., 2002; Vilcheze, et al., 2011; Xu, et al., 2011), поэтому нацеливание на гомеостаз тиолов может увеличивать или уменьшать активность INH в зависимости от механизма ингибирования.
Предлагаемый механизм для AC2P36 предполагает, что соединение обычно является тиолореактивным, а также нацелено на пулы тиолов эукариот. Эта возможность подчеркивается цитотоксическим действием AC2P36 против эукариотических макрофагов (рис. 5). Однако аналог AC2P36-J остается активным в отношении Mtb, не проявляя при этом цитотоксичности для макрофагов, что позволяет предположить, что у этого соединения могут существовать специфические функции Mtb.Кроме того, в свете известной химии, окружающей этот каркас, может быть возможно разработать ингибиторы пулов тиолов Mtb, которые действуют как пролекарство, что приводит к активации химии нацеливания на тиол только внутри бактерии. Действительно, препарат, продуцирующий оксид азота, PA-824 — это пролекарство, которое специфически способствует нитрозативному стрессу внутри бактерии (Singh, et al., 2008).
Значение
Mycobacterium tuberculosis (Mtb) должна адаптироваться к кислой среде, чтобы колонизировать и выжить в организме хозяина.Следовательно, обнаружение путей, необходимых для выживания при кислом pH и уязвимых для химического ингибирования, может выявить новые антивирулентные подходы к контролю инфекции Mtb. Здесь мы описываем небольшую молекулу AC2P36, которая избирательно убивает Mtb при кислом pH. Показано, что AC2P36 сильно истощает пулы свободных тиолов и способствует накоплению активных форм кислорода при кислом pH. AC2P36 сильно взаимодействует с антибиотиком первой линии изониазидом, подтверждая, что регулирование пулов свободных тиолов Mtb может повысить эффективность лечения Mtb.Эти данные подтверждают модель, в которой химическое истощение пулов тиолов Mtb при кислом pH приводит к повышенной чувствительности к окислительному повреждению, что приводит к уничтожению бактерий и усилению действия антибиотиков.
Экспериментальные процедуры
Бактериальные штаммы и условия роста
Mycobacterium tuberculosis (Mtb) Эксперименты, если не указано иное, проводили со штаммом Mtb CDC1551. Культуры поддерживали в стоячих колбах для тканевых культур в среде Миддлбрука 7H9 с добавлением 10% олеиновой кислоты, альбумина, декстрозы, каталазы (OADC) и 0.05% Tween-80 и инкубировали при 37 o ° C с 5% CO 2 , если не указано иное. Staphylococcus aureus. экспериментов проводили с S. aureus штамма Newman и поддерживали в пробирках Falcon на 15 мл или колбах Эрленмейера в бульоне Лурия-Бертани (LB). При необходимости среды 7H9 или LB забуферивали до pH 7,0 с помощью 100 мМ MOPS или до pH 5,7 с помощью 100 мМ MES (Piddington, et al., 2000).
EC
50 определение и спектр активности немикобактерий
Для определений EC 50 культуры инкубировали в 96-луночных микротитрационных планшетах в присутствии 8-точечного (2.5-кратное разведение AC2P36 в диапазоне от 80 мкМ до 0,13 мкМ. Культуры Mtb выращивали до средней и поздней логарифмической фазы, а затем осаждали, ресуспендировали в 7H9, забуференном при pH 7,0 или 5,7, и разливали в 96-луночные аналитические планшеты с оптической плотностью (OD) 0,1 и инкубировали в течение 6 дней. при 37 ° C с 5% CO 2 . Активность AC2P36 против немикобактерий оценивали в забуференном бульоне LB с pH 7,0 или pH 5,7, встряхивая при 200 об / мин при 37 ° C, за исключением Enterococcus faecalis, , который выращивали в среде Brain Heart Infusion в стоячих культурах в колбах при 37 ° C. С.Культуры выращивали в течение ночи и затем снова разбавляли до начальной оптической плотности (OD) 0,05 в 96-луночных микротитровальных планшетах. Были протестированы несколько грамположительных и грамотрицательных бактерий, включая: Staphylococcus aureus, штаммов Wichita (29213) или Сиэтл (25923), Escherichia coli (Migula) Castellani and Chalmers, Pseudomonas aeruginosa (Schroeter) Proteus vulgaris Hauser emend. Судебная комиссия, Enterococcus faecalis (Andrewes and Horder) Schleifer and Kilpper-Balz.Бактерии инкубировали в присутствии серии 8-точечных (2,5-кратных) разведений AC2P36 в диапазоне от 80 мкМ до 0,13 мкМ в течение 6-8 часов. Рост (OD) контролировали с использованием планшет-ридера Perkin Elmer EnVision. Культуры немикобактерий нормализовали по росту на основе контролей канамицина (100%) и ДМСО (0%), за исключением P. aeruginosa, , где тобрамицин использовали в качестве 100% положительного контроля. Значения EC 50 были рассчитаны на основе четырехпараметрической нелинейной регрессионной модели с переменным наклоном наименьших квадратов в программном пакете GraphPad Prism (вер.6).
Бактерицидная активность AC2P36
Культуры Mtb получали при OD 600 0,1 в забуференной среде 7H9 (pH 5,7) в 96-луночных планшетах, как описано выше. Культуры Mtb обрабатывали AC2P36 (в указанных концентрациях) в течение 3 дней. В конце инкубации проводили 10-кратные серийные разведения для каждого условия и подсчитывали КОЕ жизнеспособных бактерий путем посева на агар 7 ч 20 мин. Бактерицидную активность определяли путем сравнения бактериальных КОЕ после обработки с исходным инокулятом.Культуры, обработанные ДМСО и рифампицином, были включены в качестве отрицательного и положительного контролей соответственно.
Измерение внутриклеточного окислительно-восстановительного состояния
Внутриклеточное окислительно-восстановительное состояние культур Mtb, обработанных интересующими соединениями, определяли с использованием редокс-чувствительного репортерного штамма Mtb CDC1551⸬roGFP-R12 (Baker, et al., 2014; Cannon and Remington, 2006 ; Hanson, et al., 2004). Вкратце, Mtb культивировали в забуференной среде 7H9 с pH 7,0 или 5,7 без каталазы (например, OAD) в 96-луночных планшетах, содержащих 0.1-20 мкМ AC2P36 при исходном OD 600 0,2. Для каждого экспериментального условия нефлуоресцентный CDC1551 дикого типа, трансформированный пустым вектором, использовали в качестве контроля для вычитания фонового сигнала. Через 6 часов после обработки измеряли эмиссию флуоресценции при 510 нм после возбуждения при 400 и 480 нм, и измеряли относительное содержание окисленных и восстановленных форм roGFP, соответственно. Отношение 400/480 нм культур, обработанных лекарственным средством, нормализовали к контролю ДМСО, который представлял исходное окислительно-восстановительное состояние клетки.Дитиотреитол (ДТТ) использовали в качестве контроля для восстановительной среды, а диамид использовали в качестве контроля для окислительной среды.
Профилирование транскрипции и анализ данных
Для экспериментов с RNA-seq культуры Mtb выращивали при 37 ° C в вентилируемых стоячих колбах для тканевых культур Т-25 в 8 мл буферной среды 7H9 (pH 7,0 или 5,7) до OD 600 из 0.5. Затем культуры обрабатывали 10 мкМ AC2P36. В качестве базового контроля использовали культуры, обработанные равным количеством ДМСО.Каждую обработку повторяли с двумя независимыми (биологическими) повторами. После 4-часового инкубационного периода в присутствии AC2P36 общая бактериальная РНК была извлечена, проверена на качество и секвенирована, как описано ранее (Baker, et al., 2014; Rohde, et al., 2007). Данные РНК-seq анализировали с помощью программного пакета SPARTA (Johnson, et al., 2016). Гены со средним значением log 2 CPM <5 были отфильтрованы из окончательных списков дифференциальной экспрессии генов (дополнительные таблицы S2AB, S3AB, S4AB).Гены с> 2-кратной индукцией или репрессией и скорректированным p-значением (обозначенным q) <0,05 были включены в списки значительно дифференцированно регулируемых генов. Данные транскрипционного профилирования доступны в базе данных NCBI GEO (GSE89106).
Взаимосвязь структурной активности (SAR) и эукариотическая цитотоксичность AC2P36
Было закуплено несколько аналогов AC2P36 для определения взаимосвязи структурной активности. Соединения B-J (таблица 5.2.) Оценивали на ингибирующую активность в отношении Mtb с использованием 8-балльной (2.5-кратные) серии разведений в диапазоне от 80 до 0,13 мкМ при pH 5,7 аналогично определениям EC 50 , описанным выше. После 6 дней инкубации культуры анализировали на рост с использованием OD относительно рифампина (100%) и ДМСО (0%), положительного и отрицательного контроля, соответственно.
Эукариотическую цитотоксичность AC2P36 и его аналогов оценивали, как описано ранее (Johnson and Abramovitch, 2015). Вкратце, макрофаги J774 выращивали при 37 ° C (5% CO 2 ) в среде DMEM (Corning CellGro), содержащей 10% фетальной бычьей сыворотки (Thermo Scientific), 1 мМ пируват, 2 мМ L-глутамин и 1 мМ пенициллин / стрептомицин (Corning CellGro).Клетки выращивали до слияния, соскабливали и высевали в среду без антибиотиков в количестве 1 × 10 5 клеток / мл. Клеткам позволяли прикрепиться в течение ночи перед добавлением экспериментальных обработок. Клетки инкубировали в течение 3 дней при 37 ° C с 5% CO 2 и оценивали жизнеспособность с использованием набора для люциферазы CellTiter Glo (Promega). Цитотоксичность была нормализована на основе контролей 1% Triton X-100 (100%) и ДМСО (0%). Значения EC 50 и полумаксимальной клеточной цитотоксичности (CC 50 ) рассчитывали на основе четырехпараметрической нелинейной регрессионной модели с переменным наклоном в программном пакете GraphPad Prism (вер.6).
Фракционное уничтожение и усиление действия противомикробных препаратов
Культуры Mtb выращивали до средней и поздней логарифмической фазы, осаждали и ресуспендировали в буферном растворе с pH 7,0 или 5,7 7H9 без каталазы при OD 0,1. Клетки оценивали на жизнеспособность в присутствии или в отсутствие 10 мкМ AC2P36 в комбинации с: ДМСО, 1 мМ диамидом, 100 мкМ клофазимином, 0,3 мкМ рифампицином или 10 мкМ изониазидом. После 3 дней инкубации в 96-луночных микротитрационных планшетах клетки разводили серией серийных 10-кратных разведений для каждого состояния и подсчитывали КОЕ жизнеспособных бактерий, высевая на агар 7h20.
Измерение внутриклеточных активных форм кислорода и pH
Внутриклеточные активные формы кислорода измеряли с использованием реагента CellROX Green (Invitrogen), как описано ранее (Saini, et al., 2016). Вкратце, Mtb выращивали до среднего или латинского уровня в Миддлбруке 7H9 и инокулировали в 5 мл забуференного 7H9, pH 7,0 или 5,7, не содержащего каталазы, при исходной OD 0,5 и инкубировали в присутствии указанных концентраций соединений. После 4 или 24 часов инкубации к культурам добавляли конечную концентрацию 5 мкМ CellROX green (Thermo Fisher) на 1 час при 37 ° C.Клетки дважды промывали PBS с добавлением 0,05% Tween80, ресуспендировали в промывочном буфере и анализировали в соответствии с инструкциями производителя. ДТТ (20 мкМ) и диамид (2 и 20 мкМ) служили контролем. культур S. aureus выращивали в течение ночи в LB при 37 ° C со встряхиванием при 200 об / мин. Культуры осаждали и ресуспендировали в забуференной среде LB с pH 7,0 или pH 5,7 при OD 0,1. Клетки обрабатывали серией 4-точечных (2-кратных) разведений AC2P36, DTT или менадиона в диапазоне от 80 до 10 мкМ.Эквивалентный объем ДМСО добавляли в качестве отрицательного контроля. После 6 часов обработки культуры инкубировали с 5 мкМ CellROX Green в течение 30 минут при 37 ° C со встряхиванием при 200 об / мин. Клетки дважды промывали PBS и ресуспендировали в 0,6 мл PBS перед помещением в 96-луночный микротитровальный планшет. Флуоресценцию и рост анализировали с использованием планшет-ридера Perkin Elmer. Цитоплазматический pH измеряли в течение 24 часов после обработки AC2P36 при pH 5,7 по сравнению с отрицательным и положительным контролями на ДМСО или нигерицин, соответственно, с использованием метода, описанного Purdy, Niederweis и Russell (Purdy, et al., 2009).
Измерение пулов свободных тиолов
Культуры Mtb выращивали до средней и поздней логарифмической фазы в среде 7H9 без каталазы, гранулировали и повторно суспендировали в 8 мл буфера 7H9 с pH 7,0 или 5,7 при OD 0,25. Оценивали пять обработок: 1) ДМСО, 2) 2 мкМ AC2P36, 3) 20 мкМ AC2P36, 4) 20 мкМ ауранофина и 5) изониазид. Через 24 часа обработки бактерии нормализовали по OD, осаждали, дважды промывали PBS + 0,05% тилоксапола и ресуспендировали в 0,75 мл буфера для анализа тиола (100 мМ фосфат калия, pH 7.4 и 1 мМ ЭДТА). Клетки лизировали взбиванием шариков в течение 2 минут при комнатной температуре. Супернатанты удаляли и анализировали с использованием набора Cayman Thiol Detection Assay (Cayman Chemical).
Анализ образования аддукта AC2P36-GSH
В 100 мкл буфера Tris-HCl (pH 7,0 или 5,7) добавляли 80 мкМ AC2P36 вместе со 100 мкМ восстановленного глутатиона. Также проводили контроль без восстановленного глутатиона. Реакции инкубировали при комнатной температуре в течение 1 часа. Для масс-спектроскопического анализа (МС) образцы разбавляли 1: 100 водой и анализировали на масс-спектрометре Waters Xevo G2-XS QTof (Милфорд, Массачусетс, США) с отрицательным режимом ионизации электрораспылением.Параметры прибора: капиллярное напряжение 2 кВ; конус отбора проб, 40 В; температура источника 100 ° C; температура десольватации 350 ° С; конусный расход газа 25 л / ч; расход десольватационного газа 600 л / ч. Хроматографическое разделение выполняли в системе ультраэффективного жидкостного хроматографа Waters (ACQUITY UPLC). Растворителями были (A) вода и (B) ацетонитрил. Скорость потока составляла 0,2 мл / мин с градиентом A / B = 50/50 в течение 2 минут. Диапазон получаемых масс составлял 50–1 500 Да. Эксперимент был повторен по крайней мере с двумя биологическими повторами с аналогичными результатами.
Вклад авторов
GBC, BKJ, NDH и RBA разработали эксперименты; GBC, BKJ, CJC, RJF, HZ и EH проводили эксперименты; GBC, BKJ и RBA написали рукопись.
Коды доступа
Данные транскрипционного профилирования были отправлены в базу данных NCBI GEO (инвентарный номер GSE89106).
Дополнительный рисунок 1. AC2P36 не влияет на pH цитоплазмы Mtb при pH 5,7. ДМСО и нигерицин добавляют в качестве отрицательного и положительного контролей. Данные представляют два независимых эксперимента.Дополнительная таблица 1.
AC2P36 спектр активности против грамположительных и грамотрицательных бактерий.
Дополнительная таблица 2. Профилирование транскрипции Mtb, обработанного ДМСО, по сравнению с AC2P36 при pH 7,0.
Дополнительная таблица 3. Профилирование транскрипции Mtb, обработанного ДМСО, по сравнению с AC2P36 при pH 5,7.
Дополнительная таблица 4. Транскрипционное профилирование Mtb, обработанного ДМСО, при pH 5,7 по сравнению с pH 7,0.
Благодарности
Мы благодарим Региональный центр передового опыта Новой Англии (U54 AI057159) за предоставление скрининговых библиотек, а также Су Чанга и Дуга Флуда за помощь в подготовке библиотек соединений для скрининга.