О перечне медицинских показаний для санаторно-курортного лечения детского населения в КОГБУЗ «Санаторий для детей с родителями «Лесная сказка»
В связи с использованием минеральной воды для бальнеологического лечения КОГБУЗ «Санаторий для детей с родителями «Лесная сказка», на санаторно-курортное лечение направляются несовершеннолетние с болезнями органов дыхания, болезнями органов пищеварения, болезнями мочеполовой системы.
Медицинские показания для санаторно-курортного лечения детского населения (в соответствии с приказом № 281н от 05.05. 2016 г. Минздрава России, приложение №2)
VIII раздел. Медицинские показания для санаторно-курортного лечения детского населения с болезнями органов дыхания:
- Вирусная пневмония, не классифицированная в других рубриках (МКБ-10 — J 12).
- Пневмония, вызванная Streptococcus pneumoniae (МКБ-10 — J 13).
- Пневмония, вызванная Haemophilus influenza (МКБ-10 — J 14).
- Бактериальная пневмония, не классифицированная в других рубриках (МКБ-10 — J 15).

- Пневмония при болезнях, классифицированных в других рубриках (МКБ-10 — J 17). В стадии реконвалесценции (не ранее 4 недель от начала заболевания).
- Простой и слизисто-гнойный хронический бронхит (МКБ-10 — J 41).
- Эмфизема (МКБ-10 — J 43).
- Другая хроническая обструктивная легочная болезнь (МКБ-10 — J 44).
- Другая уточненная хроническая обструктивная легочная болезнь (МКБ-10 — J 44.8).
- Астма (МКБ-10 — J 45).

IX раздел. Медицинские показания для санаторно-курортного лечения детского населения с болезнями органов пищеварения:
- Гастрит и дуоденит (МКБ-10 — К 29), легкой, средней степени тяжести в стадии ремиссии.
- Другие функциональные кишечные нарушения (МКБ-10 — К 59), средней степени тяжести.
- Запор (МКБ-10 — К 59.0), средней степени тяжести.
- Другие уточненные болезни желчного пузыря (МКБ-10 — К 82.8), дискинезия пузырного протока, желчного пузыря.
XII раздел. Перечень медицинских показаний для санаторно-курортного лечения детского населения с болезнями мочеполовой системы:
- Хронический обструктивный пиелонефрит (МКБ-10 -N11. 1),вне рецидива.
- Цистит (МКБ-10 — N30) в стадии ремиссии.
- Интерстициальный цистит (хронический) (МКБ-10 -N30.1), в стадии ремиссии
- Другой хронический цистит (МКБ-10 — N30.2),в стадии ремиссии.
 1),вне рецидива.
1),вне рецидива.В связи с уменьшением срока оздоровительного лечения до 19 дней обучение детей школьного возраста по программам начального общего, основного общего, среднего общего образования осуществляться не будет. (Письмо Министерства образования и науки Российской Федерации от 27.05.2016 ВК- 1179/07 «О дополнительных разъяснениях».
Запись опубликована автором Редактор в рубрике Новости санатория.
Нозокомиальная пневмония — МКБ-10 | Medum.ru
Ниже приведён список действующих веществ, относящихся
к коду J18 МКБ-10 (наименования фармакологических групп и
перечень торговых названий, связанных с этим кодом).
Фармакологическая группа: Макролиды и азалиды
Фармакологическая группа: Монобактамы
Фармакологическая группа: Секретолитики и стимуляторы моторной функции дыхательных путей
Фармакологические группы: Секретолитики и стимуляторы моторной функции дыхательных путей, Прочие ненаркотические анальгетики, включая нестероидные и другие противовоспалительные средства
Фармакологическая группа: Секретолитики и стимуляторы моторной функции дыхательных путей
Фармакологическая группа: Аминогликозиды
Фармакологические группы: Диуретики, Секретолитики и стимуляторы моторной функции дыхательных путей
Фармакологическая группа: Пенициллины
Фармакологическая группа: Пенициллины в комбинации с другими препаратами
Фармакологическая группа: Пенициллины
Фармакологическая группа: Пенициллины в комбинации с другими препаратами
Фармакологическая группа: Пенициллины в комбинации с другими препаратами
Фармакологические группы: Секретолитики и стимуляторы моторной функции дыхательных путей, Детоксицирующие средства, включая антидоты
Фармакологическая группа: Пенициллины
Фармакологическая группа: Средства, нормализующие микрофлору кишечника
Фармакологическая группа: Средства, нормализующие микрофлору кишечника, в комбинации с другими препаратами
Фармакологическая группа: Секретолитики и стимуляторы моторной функции дыхательных путей
Фармакологическая группа: Секретолитики и стимуляторы моторной функции дыхательных путей в комбинации с другими препаратами
Фармакологическая группа: Секретолитики и стимуляторы моторной функции дыхательных путей в комбинации с другими препаратами
Фармакологическая группа: Секретолитики и стимуляторы моторной функции дыхательных путей в комбинации с другими препаратами
Фармакологическая группа: Гликопептиды
Фармакологические группы: Аминогликозиды, Офтальмологические препараты
Фармакологическая группа: Противокашлевые препараты
Фармакологические группы: Адрено- и симпатомиметики (альфа-, бета-) в комбинации с другими препаратами, Противокашлевые средства в комбинации с другими препаратами
Фармакологические группы: Адрено- и симпатомиметики (альфа-, бета-) в комбинации с другими препаратами, Противокашлевые средства в комбинации с другими препаратами
Фармакологическая группа: Хинолоны/фторхинолоны
Фармакологическая группа: Макролиды и азалиды
Фармакологическая группа: Макролиды и азалиды
Фармакологические группы: Антигипоксанты и антиоксиданты, Витамины и витаминоподобные средства
Фармакологические группы: Антигипоксанты и антиоксиданты, Антисептики и дезинфицирующие средства, Регуляторы водно-электролитного баланса и КЩС
Фармакологическая группа: Макролиды и азалиды
Фармакологическая группа: Тетрациклины
Фармакологическая группа: Карбапенемы
Фармакологическая группа: Секретолитики и стимуляторы моторной функции дыхательных путей
Фармакологическая группа: НПВС — Производные пропионовой кислоты
Фармакологическая группа: Интерлейкины
Фармакологическая группа: Макро- и микроэлементы
Фармакологическая группа: Аминогликозиды
Фармакологическая группа: Пенициллины
Фармакологическая группа: Секретолитики и стимуляторы моторной функции дыхательных путей
Фармакологическая группа: Макролиды и азалиды
Фармакологическая группа: Линкозамиды
Фармакологическая группа: Сульфаниламиды
Фармакологические группы: Офтальмологические препараты, Хинолоны/фторхинолоны
Фармакологическая группа: Индукторы интерферонов
Фармакологическая группа: Карбапенемы
Фармакологическая группа: Тетрациклины
Фармакологические группы: Прочие иммуномодуляторы, Противовирусные средства (исключая ВИЧ)
Фармакологическая группа: Прочие синтетические антибактериальные средства
Фармакологическая группа: Макролиды и азалиды
Фармакологическая группа: Прочие иммуномодуляторы
Фармакологическая группа: Аминогликозиды
Фармакологическая группа: Противокашлевые препараты
Фармакологическая группа: Прочие иммуномодуляторы
Фармакологическая группа: Макролиды и азалиды
Фармакологические группы: Макролиды и азалиды в комбинации с другими препаратами, Тетрациклины в комбинации с другими препаратами
Фармакологические группы: Офтальмологические препараты, Хинолоны/фторхинолоны
Фармакологическая группа: Вакцины, сыворотки, фаги и анатоксины
Фармакологическая группа: Пенициллины
Фармакологическая группа: Антигипоксанты и антиоксиданты
Фармакологическая группа: Прочие антибиотики
Фармакологическая группа: Витамины и витаминоподобные средства
Фармакологическая группа: Макролиды и азалиды
Фармакологическая группа: Аминогликозиды
Фармакологическая группа: Хинолоны/фторхинолоны
Фармакологическая группа: Макролиды и азалиды
Фармакологическая группа: Пенициллины
Фармакологическая группа: Сульфаниламиды
Фармакологическая группа: Сульфаниламиды
Фармакологическая группа: Сульфаниламиды
Фармакологические группы: Сульфаниламиды в комбинации с другими препаратами, Прочие синтетические антибактериальные средства в комбинации с другими препаратами
Фармакологическая группа: Сульфаниламиды
Фармакологические группы: Офтальмологические препараты, Сульфаниламиды
Фармакологическая группа: Сульфаниламиды
Фармакологическая группа: Гликопептиды
Фармакологическая группа: Тетрациклины
Фармакологическая группа: Пенициллины в комбинации с другими препаратами
Фармакологическая группа: Прочие синтетические антибактериальные средства
Фармакологические группы: Прочие синтетические антибактериальные средства в комбинации с другими препаратами, Хинолоны/фторхинолоны в комбинации с другими препаратами
Фармакологические группы: Аминогликозиды, Офтальмологические препараты
Фармакологическая группа: Ферменты и антиферменты
Фармакологическая группа: м-Холинолитики
Фармакологическая группа: Пенициллины
Фармакологическая группа: Прочие синтетические антибактериальные средства
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины в комбинации с другими препаратами
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Цефалоспорины в комбинации с другими препаратами
Фармакологическая группа: Цефалоспорины
Фармакологическая группа: Антигипоксанты и антиоксиданты
Фармакологические группы: Прочие противомикробные, противопаразитарные и противоглистные средства, Секретолитики и стимуляторы моторной функции дыхательных путей
Фармакологическая группа: Секретолитики и стимуляторы моторной функции дыхательных путей
Фармакологические группы: Макролиды и азалиды, Офтальмологические препараты
Фармакологическая группа: Прочие иммуномодуляторы
Фармакологическая группа: Прочие иммуномодуляторы
Фармакологические группы: Антисептики и дезинфицирующие средства, Прочие иммуномодуляторы
Фармакологическая группа: Индукторы интерферонов
Фармакологическая группа: Цефалоспорины в комбинации с другими препаратами
В КР объединяют данные по COVID-19 и пневмонии.
 Пересмотрят ли статистику по смертности?
Пересмотрят ли статистику по смертности?
Всего за время эпидемии COVID-19 в Кыргызстане от коронавируса скончались 167 человек, от внебольничной пневмонии – 612. В социальных сетях кыргызстанского сегмента появляются различные сообщения о том, что из-за отсутствия мест в больницах многие граждане лечатся на дому и умирают.
Ранее группа медиков и ученых страны заявили о необходимости лечить всех, кто заболел внебольничной пневмонией, но при этом их результаты на коронавирус вышли отрицательными, как заразившихся COVID-19. Но до последнего дня Республиканский оперативный штаб разделял эти цифры и вел отдельный учет по каждому из этих диагнозов.
А 16 июля вице-премьер-министр Аида Исмаилова заявила, что Минздрав издал приказ, согласно которому случаи внебольничной пневмонии считаются проявлением COVID-19.
Советник Министерства здравоохранения по доказательной медицине Бермет Барыктабасова в интервью «Азаттыку» 16 июля рассказала о важности того, чтобы заболевших пневмонией рассматривали как коронавирусных больных:
Бермет Барыктабасова.
– С самого начала я говорила, что все умершие за последние три недели – это заразившиеся COVID-19. А в марте я отмечала, что тесты имеют погрешность, то есть, они могут быть ложно-отрицательными. Все ведь зависит от технологии забора. Если взяли неправильно или неправильно определили, то человеку ставят отрицательный результат, как будто у него нет коронавируса. А он умирает.
И вот когда он доходит до патологоанатомов, они уже могут исследовать более глубоко — взять смывы с бронхолегочного дерева. Недавно они взяли анализы у всех умерших, чьи анализы на COVID-19 были отрицательными и которым ставили внебольничную пневмонию. У всех подтвердился коронавирус. То есть внебольничной пневмонии нет. Есть неподтвержденный ковид.
– Что значит «неподтвержденный ковид»?
– Недавно Минздрав утвердил третью версию национального клинического руководства по COVID-19. Там в самом начале написано, как ставится диагноз. Есть международная классификация болезней 10 пересмотра (МКБ-10), и в нее именно про коронавирус добавили три пункта.
Первое – это подтвержденный COVID-19, то есть коронавирусная инфекция (подтвержденная), когда результат ПЦР-теста положительный. А второй шифр значится как неподтвержденная коронавирусная инфекция , то есть ПЦР-тест вышел отрицательным. Но это COVID-19, просто не подтвержденный лабораторно, Вот и все.
Его может подтвердить рентген, например. Или компьютерная томография, клиническая картина, эпиданамнез. Может подтвердить вирусология, посмертно. Это то, что сейчас делают патологоанатомы. Они и судмедэксперты как раз на вскрытии видят классическую картину описания вот коронавирусной инфекции.
Вот эти разночтения в статистике — они же пошли с самого начала. Хотя точно видно, что симптомы одни и те же: к примеру, 10 дней температура, одышка, человек задыхается, слабеет, умирает… Но ему ставят диагноз, что «нет, это не COVID-19, у него был сахарный диабет или сердечная недостаточность, или пневмония». И сколько таких диагнозов — «пневмония» — было поставлено?
С самых первых дней это было, еще когда в Сузакском районе пошли первые случаи смерти. Там умер один медработник, психиатр. Он ходил в обсервацию, осматривал контактных лиц – искал, выявлял, брал анализы… Потом сам заболел. И налицо была абсолютно классическая картина COVID-19. Причем очень быстро болезнь развивалась. Пять дней он был подключен к ИВЛ. Умер. Но ему не поставили коронавирус, быстро похоронили без вскрытия и сказали, что умер от пневмонии.
Там умер один медработник, психиатр. Он ходил в обсервацию, осматривал контактных лиц – искал, выявлял, брал анализы… Потом сам заболел. И налицо была абсолютно классическая картина COVID-19. Причем очень быстро болезнь развивалась. Пять дней он был подключен к ИВЛ. Умер. Но ему не поставили коронавирус, быстро похоронили без вскрытия и сказали, что умер от пневмонии.
– Вы говорите, что изначально предлагали рассматривать пневмонию как коронавирус. А до этого открытого обращения вы пытались хотя бы устно сказать об этом Минздраву или Республиканскому штабу?
– Мы увидели, что несмотря на то, что давали Минздраву эти рекомендации, они все равно продолжают разделять пневмонию и COVID-19. Я например с патологоанатомами разговаривала, наверное, где-то две недели назад.
– Вы предложили рассматривать пневмонию как COVID-19, опираясь на научные доводы, или только из-за того, что симптомы заболеваний схожи?
– Несколько дней назад патологоанатом сообщила, что они проверили тела 43 умерших, которым при жизни ставили отрицательный результат по коронавирусу. Но взятые патологоанатомами анализы показали у всех умерших положительный результат на COVID-19. Поэтому статистику уже можно не делить. Потому что внебольничная пневмония лечится совершенно по-другому.
Но взятые патологоанатомами анализы показали у всех умерших положительный результат на COVID-19. Поэтому статистику уже можно не делить. Потому что внебольничная пневмония лечится совершенно по-другому.
– А как она лечится?
– Ну, смотрите. Внебольничная пневмония чаще носит не вирусную, а бактериальную форму. И там антибиотики назначаются с первых дней. Если же поставили COVID-19, то лечение другое, госпитализация другая, вскрытие совсем другое, хоронят совсем по-другому… Это совершенно другая инфекция и другая болезнь.
Но если пишут, что внебольничная пневмония, хоронят как обычно, лечат как обычно тогда как антибиотиками коронавирус совсем не лечат… Мы должны добиться, чтобы это заблуждение, вот это недоразумение, вся эта ошибочная постановка диагноза прекратились. Мы не должны никого обманывать. Все сейчас понимают, что это COVID-19.
– Вся эта неразбериха говорит о слабости кыргызстанской медицины или халатности ответственных руководителей?
– Самое главное тут, самая большая загвоздка – это ошибка системы. Фонд обязательного медицинского страхования (ФОМС) принимал вот этот диагноз — «внебольничная пневмония», а «неподтвержденный COVID-19» не принимал. И поэтому все медики записывали такие смертельные случаи под шифром внебольничной пневмонии. А это совсем разные шифры! Поэтому все диагнозы прятались под внебольничной пневмонией.
Фонд обязательного медицинского страхования (ФОМС) принимал вот этот диагноз — «внебольничная пневмония», а «неподтвержденный COVID-19» не принимал. И поэтому все медики записывали такие смертельные случаи под шифром внебольничной пневмонии. А это совсем разные шифры! Поэтому все диагнозы прятались под внебольничной пневмонией.
Но сегодня все цифры смертности нужно заново пересмотреть. Пересмотреть и все случаи внебольничной пневмонии, все проверить и добавить их к этой коронавирусной инфекции, потому что других смертей просто нет. У нас годами не было смертности от внебольничной пневмонии, от гриппа, от других ОРВИ — просто не было. Смертность была от кори, когда в стране были вспышки – в 2017, 2018 и 2019 годах. И тогда смертность от кори была очень высокой, но думаю смертность от коронавирусной инфекции теперь ее догонит.
NO
Перевод с кыргызского. Оригинал статьи здесь.
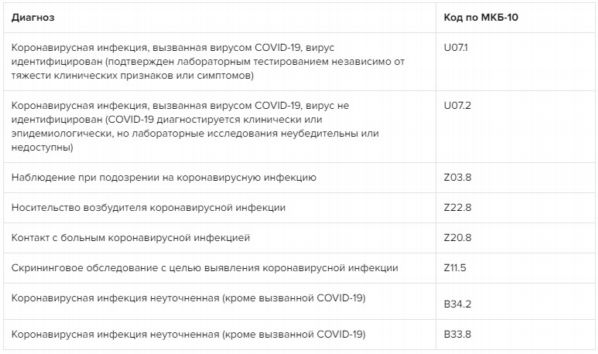
МКБ-10 Международная классификация болезней 10-го пересмотра || Комитет солдатских матерей России.
 Болезни органов дыхания (J00-J99). Грипп и пневмония
Болезни органов дыхания (J00-J99). Грипп и пневмония
J09 Грипп, вызванный выявленным вирусом зоонозного или пандемического гриппа
Примечание. Для использования этой категории следует обратиться к руководящим принципам Глобальной программы по гриппу ВОЗ (GIP, www.who.int/influenza/)
Грипп, вызванный штаммами вируса гриппа, имеющих особое эпидемиологическое значение, с передачей животными и человеком
При необходимости используйте дополнительный код, чтобы идентифицировать пневмонию или другие проявления.
Исключено:
Haemophilus influenzae [H. influenzae]:
- инфекция БДУ (A49.2)
- менингит (G00.0)
- пневмония (J14)
грипп, с выявленным вирусом сезонного гриппа (J10.-)
J10 Грипп, вызванный идентифицированным вирусом сезонного гриппа
Включено: грипп, вызванный идентифицированным вирусом гриппа B или C
Исключены:
вызванная(ый) Haemophilus influenzae [палочкой Афанасьева-Пфейффера]:
- инфекция БДУ (A49. 2)
- менингит (G00.0)
- пневмония (J14)
 2)
2)
грипп, вызванный выявленным вирусом зоонозного или пандемического гриппа (J09)
J10.0 Грипп с пневмонией, сезонный вирус гриппа идентифицирован
Гриппозная (бронхо)пневмония, сезонный вирус гриппа идентифицирован
J10.1 Грипп с другими респираторными проявлениями, сезонный вирус гриппа идентифицирован
Грипп, сезонный вирус гриппа идентифицирован
Гриппозная(ый):
- острая респираторная инфекция верхних дыхательных путей, сезонный вирус гриппа идентифицирован
- ларингит, сезонный вирус гриппа идентифицирован
- фарингит, сезонный вирус гриппа идентифицирован
- плевральный выпот, сезонный вирус гриппа идентифицирован
J10.8 Грипп с другими проявлениями, сезонный вирус гриппа идентифицирован
Энцефалопатия, вызванная гриппом, сезонный вирус гриппа идентифицирован
Гриппозный:
- гастроэнтерит, сезонный вирус гриппа идентифицирован
- иокардит (острый), сезонный вирус гриппа идентифицирован
J11 Грипп, вирус не идентифицирован
Включены:
- грипп, упоминание об идентификации вируса отсутствует
- вирусный грипп, упоминание об идентификации вируса отсутствует
Исключены: вызванная(ый) Haemophilus influenzae [палочкой Афанасьева-Пфейффера]:
- инфекция БДУ (A49. 2)
- менингит (G00.0)
- пневмония (J14)
 2)
2)
J11.0 Грипп с пневмонией, вирус не идентифицирован
Гриппозная (бронхо)пневмония неуточненная или без упоминания об идентификации вируса
J11.1 Грипп с другими респираторными проявлениями, вирус не идентифицирован
Грипп БДУ
Гриппозная(ый):
острая респираторная инфекция неуточненный(ая) верхних дыхательных путей или вирус
J11.8 Грипп с другими проявлениями, вирус не идентифицирован
Энцефалопатия, вызванная гриппом
J12 Вирусная пневмония, не классифицированная в других рубриках
Включена: бронхопневмония, вызванная другими вирусами, отличными от вируса гриппа
Исключены:
- врожденный краснушный пневмонит (P35.0)
- пневмония:
- аспирационная:
- БДУ (J69.0)
- при анестезии:
- во время родов и родоразрешения (O74.0)
- во время беременности (O29. 0)
- в послеродовом периоде (O89.0)
- новорожденного (P24.9)
- при вдыхании твердых и жидких веществ (J69.-)
- при гриппе (J09, J10.0, J11.0)
- интерстициальная БДУ (J84.9)
- жировая (J69.1)
- вирусная врожденная (P23.0)
- тяжелый острый респираторный синдром [SARS] (U04.9)
 0)
0)
J12.0 Аденовирусная пневмония
J12.1Пневмония, вызваннаяреспираторным синцитиальным вирусом
J12.2Пневмония, вызванная вирусом парагриппа
J12.3 Пневмония, вызванная человеческим метапневмовирусом
J12.8 Другая вирусная пневмония
J12.9 Вирусная пневмония неуточненная
J13 Пневмония, вызванная Streptococcus pneumoniae
Бронхопневмония, вызванная S. pneumoniae
Исключены:
- врожденная пневмония, вызванная S.pneumoniae (P23.6)
- пневмония, вызванная другими стрептококками (J15.3-J15.4)
J14 Пневмония, вызванная Haemophilus influenzae [палочкой Афанасьева-Пфейффера]
Бронхопневмония, вызванная H. influenzae
influenzae
Исключена: врожденная пневмония, вызванная H.influenzae (P23.6)
J15 Бактериальная пневмония, не классифицированная в других рубриках
Включена: бронхопневмония, вызванная другими, отличными от S.pneumoniae и H.influenzae бактериями
Исключены:
- пневмония, вызванная хламидиями (J16.0)
- врожденная пневмония (P23.-)
- болезнь легионеров (A48.1)
J15.0 Пневмония, вызванная Klebsiella pneumoniae
J15.1 Пневмония, вызванная Pseudomonas (синегнойной палочкой)
J15.2 Пневмония, вызванная стафилококком
J15.3 Пневмония, вызванная стрептококком группы B
J15.4 Пневмония, вызванная другими стрептококками
Исключены: пневмония, вызванная:
- стрептококком группы B (J15.3)
- Streptococcus pneumoniae (J13)
J15.5 Пневмония, вызванная Escherichia coli
J15.6 Пневмония, вызванная другими грамотрицательными бактериями
Пневмония, вызванная:
- грамотрицательными (аэробными) бактериями БДУ
- Serratia marcescens
J15. 7 Пневмония, вызванная Mycoplasma pneumoniae
7 Пневмония, вызванная Mycoplasma pneumoniae
J15.8 Другие бактериальные пневмонии
J15.9 Бактериальная пневмония неуточненная
J16 Пневмония, вызванная другими инфекционными возбудителями, не классифицированная в других рубриках
Исключены:
- орнитоз (A70)
- пневмоцистоз B59
- пневмония:
- БДУ (J18.9)
- врожденная (P23.-)
J16.0 Пневмония, вызванная хламидиями
J16.8 Пневмония, вызванная другими уточненными инфекционными возбудителями
J17* Пневмония при болезнях, классифицированных в других рубриках
J17.0* Пневмония при бактериальных болезнях, классифицированных в других рубриках
Пневмония при:
- актиномикозе (A42.0*)
- сибирской язве (A22.1*)
- гонорее (A54.8*)
- нокардиозе (A43.0*)
- сальмонеллезе (A02.2*)
- туляремии (A21.2*)
- брюшном тифе (A01.0*)
- коклюше (A37. -*)
 -*)
-*)
J17.1* Пневмония при вирусных болезнях, классифицированных в других рубриках
Пневмония при:
- цитомегаловирусной болезни (B25.0*)
- кори (B05.2*)
- краснухе (B06.8*)
- ветряной оспе (B01.2*)
J17.2* Пневмония при микозах
Пневмония при:
- аспергиллезе (B44.0-B44.1*)
- кандидозе (B37.1*)
- кокцидиоидомикозе (B38.0-B38.2*)
- гистоплазмозе (B39.-*)
J17.3* Пневмония при паразитарных болезнях
Пневмония при:
- аскаридозе (B77.8*)
- шистосомозе (B65.-*)
- токсоплазмозе (B58.3*)
J17.8* Пневмония при других болезнях, классифицированных в других рубриках
Пневмония при:
- орнитозе (A70*)
- лихорадке Ку (A78*)
- ревматической лихорадке (I00*)
- спирохетозе, не классифицированная в других рубриках (A69.8*)
J18 Пневмония без уточнения возбудителя
Исключены:
абсцесс легкого с пневмонией (J85. 1)
1)
лекарственные интерстициальные болезни легкого (J70.2-J70.4)
пневмония:
аспирационная:
БДУ (J69.0)
при анестезии:
- во время родов и родоразрешения (O74.0)
- во время беременности (O29.0)
- в послеродовом периоде (O89.0)
новорожденного (P24.9)
при вдыхании твердых и жидких веществ (J69.-)
врожденная (P23.9)
интерстициальная БДУ (J84.9)
жировая (J69.1)
обычная интерстициальная (J84.1)
пневмонит, вызванный внешними агентами (J67-J70)
J18.0 Бронхопневмония неуточненная
Исключен: бронхиолит (J21.-)
J18.1 Долевая пневмония неуточненная
J18.2 Гипостатическая пневмония неуточненная
J18.8 Другая пневмония, возбудитель не уточнен
J18.9 Пневмония неуточненная
Класс болезней по МКБ-10 | Код заболевания по МКБ-10 | Наименование заболевания |
Болезни эндокринной системы, расстройства питания и нарушения обмена веществ (класс IV по МКБ-10) | Е66 | Ожирение |
Е66. | Ожирение, обусловленное избыточным поступлением энергетических ресурсов | |
Болезни системы кровообращения (класс IX по МКБ-10) | I10 | Эссенциальная (первичная) гипертензия |
I34 | Неревматические поражения митрального клапана | |
I34.0 | Митральная (клапанная) недостаточность (не более I степени) | |
I34.8 | Другие неревматические поражения митрального клапана | |
I35 | Неревматические поражения аортального клапана | |
I35. | Аортальная (клапанная) недостаточность (не более I степени) | |
I35.8 | Другие поражения аортального клапана | |
Болезни органов дыхания (класс X по МКБ-10) | J12 | Вирусная пневмония, не классифицированная в других рубриках |
J13 | Пневмония, вызванная Streptococcus pneumoniae | |
J14 | Пневмония, вызванная Haemophilus influenza [палочкой Афанасьева-Пфейффера] | |
J15 | Бактериальная пневмония, не классифицированная в других рубриках | |
J15. | Пневмония, вызванная Klebsiella pneumonia | |
J15.2 | Пневмония, вызванная стафилококком | |
J15.3 | Пневмония, вызванная стрептококком группы В | |
J15.4 | Пневмония, вызванная другими стрептококками | |
J15.5 | Пневмония, вызванная Escherichia coli | |
J15.6 | Пневмония, вызванная другими аэробными грамотрицательными бактериями | |
J15.7 | Пневмония, вызванная Mycoplasma pneumonia | |
J15. | Другие бактериальные пневмонии | |
J17 | Пневмония при болезнях, классифицированных в других рубриках | |
J17.0 | Пневмония при бактериальных болезнях, классифицированных в других рубриках | |
J17.2 | Пневмония при микозах | |
J17.3 | Пневмония при паразитарных болезнях | |
J41 | Простой и слизисто-гнойный хронический бронхит | |
J43 | Эмфизема | |
J43.0 | Синдром Мак-Леода | |
J44 | Другая хроническая обструктивная легочная болезнь | |
J44. | Другая уточненная хроническая обструктивная легочная болезнь | |
Болезни органов пищеварения (класс XI по МКБ-10) | К20 | Эзофагит |
К21 | Гастроэзофагеальный рефлюкс | |
К21.0 | Гастроэзофагеальный рефлюкс с эзофагитом | |
К21.9 | Гастроэзофагеальный рефлюкс без эзофагита | |
К22 | Другие болезни пищевода | |
К22.0 | Ахалазия кардиальной части | |
К22. | Язва пищевода | |
К25 | Язва желудка | |
К26 | Язва двенадцатиперстной кишки | |
К29 | Гастрит и дуоденит | |
К29.8 | Дуоденит | |
К52 | Другие неинфекционные гастроэнтериты и колиты | |
К52.2 | Аллергический и алиментарный гастроэнтерит и колит | |
К58 | Синдром раздраженного кишечника | |
К58. | Синдром раздраженного кишечника без диареи | |
К59 | Другие функциональные кишечные нарушения | |
К59.0 | Запор | |
К80 | Желчнокаменная болезнь [холелитиаз] | |
К80.1 | Камни желчного пузыря с другим холециститом | |
К80.2 | Камни желчного пузыря без холецистита | |
К80.5 | Камни желчного протока без холангита или холецистита | |
К81 | Холецистит | |
К81. | Хронический холецистит | |
К82 | Другие болезни желчного пузыря | |
К82.8 | Другие уточненные болезни желчного пузыря | |
К90 | Нарушения всасывания в кишечнике | |
К90.0 | Целиакия | |
К91.5 | Постхолецистэктомический синдром | |
Болезни мочеполовой системы (класс XIV по МКБ-10) | N70 | Сальпингит и оофорит |
N70.1 | Хронический сальпингит и оофорит | |
N71 | Воспалительные болезни матки, кроме шейки матки | |
N71. | Хронические воспалительные болезни матки | |
N76 | Другие воспалительные болезни влагалища и вульвы | |
N76.1 | Подострый и хронический вагинит | |
N91 | Отсутствие менструаций, скудные или редкие менструации | |
N94 | Болевые и другие состояния, связанные с женскими половыми органами и менструальным циклом | |
N94.0 | Боли в середине менструального цикла | |
N94.3 | Синдром предменструального напряжения | |
N94. | Первичная дисменорея | |
N94.5 | Вторичная дисменорея |
 0
0 1
1 0
0 8
8 8
8 1
1 9
9 1
1 1
1 4
4Ломоносовская межрайонная больница им. И.Н.Юдченко
Дорогие друзья!
Мы рады приветствовать Вас на официальном сайте Государственного бюджетного учреждения здравоохранения Ленинградской области «Ломоносовская межрайонная больница им. И.Н.Юдченко».
Наша больница оказывает высококвалифицированную медицинскую помощь уже более 140 лет.
В составе ГБУЗ ЛО «Ломоносовская МБ» 6 стационарных круглосуточных отделений, отделение скорой медицинской помощи диагностические подразделения, клинико-диагностическая лаборатория, стоматологическая поликлиника, больница сестринского ухода, 2 участковые больницы различного профиля, консультативная поликлиника, развитая сеть первичной медико-санитарной и специализированной медицинской помощи, обеспеченную высококвалифицированными кадрами.
В больнице работает коллектив со сложившимися традициями из 850 человек, из них более 160 врачей, 330 специалистов из числа среднего медицинского персонала.
На нашем сайте можно получить информацию об истории больницы, ее структуре, профиле работы, сотрудниках учреждения, оказываемых платных медицинских услугах и многом другом. Искренне надеемся, что наш сайт станет вашим надежным помощником!
Ждем от вас вопросов по организации работы нашей больницы. Вы также можете присылать свои замечания и предложения, которые, на ваш взгляд, будут способствовать улучшению лечебного процесса.
Наши телефоны:
Консультативная поликлиника: (г.Ломоносов, ул.Александровская, д. 30 литА)
Регистратура поликлиники: +7-812-423-07-18, колл-центр: +7-812-339-60-77.
Касса платных услуг: +7-812-423-09-70.
Детское поликлиническое отделение: +7-991-02-72-953 (г. Ломоносов, Иликовский пр., д. 1/3 лит.А)
Стоматологическая поликлиника: +7-953-372-84-14, колл-центр +7-812-339-60-77 (г. Ломоносов, ул.Еленинская, д.13 лит И)
Ломоносов, ул.Еленинская, д.13 лит И)
Больница: (г.Ломоносов, ул.Еленинская, д. 13 / ул.Михайловская, д. 2)
Стол справок: +7-812-679-29-54, +7-991-027-29-69.
Регистратура стоматологической поликлиники: 8-953-372-84-14, колл-центр: +7-812-339-60-77.
Касса платных услуг больницы: +7-812-423-04-00.
Приёмное отделение:+7-812-423-07-69, +7-813-765-26-11, +7-812-679-47-90
Гинекологическое отделение: +7-953-372-44-57, +7-953-372-42-41
ФГДС +7-991-032-40-94
УЗИ +7-991-032-40-91
Горячая линия для обращения граждан: понедельник – пятница с 9.00 – 17.00 по номеру: +7-812-423-00-12 , +7-953-372-43-88 (круглосуточно).
Для информации:
Скорая помощь: +7-812-423-06-71, на территории Ломоносовского района : 03
Телефон регистратуры консультативной поликлиники на ул. Александровской д. 30 : +7-812-423-07-18
Гинекологическое отделение , номер мобильный: +7-953-372-44-57, +7-953-372-42-41
ПРОТИВОТУБЕРКУЛЕЗНЫЙ КАБИНЕТ: кабинет врача-фтизиатра взрослого:+7-812-679-29-54 добавочный 2146 — г. Ломоносов, ул.Еленинская, д.13
Ломоносов, ул.Еленинская, д.13
Произошла ошибка при настройке вашего пользовательского файла cookie
Произошла ошибка при настройке вашего пользовательского файла cookie
Этот сайт использует файлы cookie для повышения производительности. Если ваш браузер не принимает файлы cookie, вы не можете просматривать этот сайт.
Настройка вашего браузера для приема файлов cookie
Существует множество причин, по которым cookie не может быть установлен правильно. Ниже приведены наиболее частые причины:
- В вашем браузере отключены файлы cookie.Вам необходимо сбросить настройки вашего браузера, чтобы он принимал файлы cookie, или чтобы спросить вас, хотите ли вы принимать файлы cookie.
- Ваш браузер спрашивает вас, хотите ли вы принимать файлы cookie, и вы отказались.
Чтобы принять файлы cookie с этого сайта, используйте кнопку «Назад» и примите файлы cookie. - Ваш браузер не поддерживает файлы cookie. Если вы подозреваете это, попробуйте другой браузер.
- Дата на вашем компьютере в прошлом. Если часы вашего компьютера показывают дату до 1 января 1970 г.,
браузер автоматически забудет файл cookie.Чтобы исправить это, установите правильное время и дату на своем компьютере. - Вы установили приложение, которое отслеживает или блокирует установку файлов cookie.
Вы должны отключить приложение при входе в систему или проконсультироваться с системным администратором.

Почему этому сайту требуются файлы cookie?
Этот сайт использует файлы cookie для повышения производительности, запоминая, что вы вошли в систему, когда переходите со страницы на страницу.Чтобы предоставить доступ без файлов cookie
потребует, чтобы сайт создавал новый сеанс для каждой посещаемой страницы, что замедляет работу системы до неприемлемого уровня.
Что сохраняется в файле cookie?
Этот сайт не хранит ничего, кроме автоматически сгенерированного идентификатора сеанса в cookie; никакая другая информация не фиксируется.
Как правило, в cookie-файлах может храниться только информация, которую вы предоставляете, или выбор, который вы делаете при посещении веб-сайта.Например, сайт
не может определить ваше имя электронной почты, пока вы не введете его. Разрешение веб-сайту создавать файлы cookie не дает этому или любому другому сайту доступа к
остальной части вашего компьютера, и только сайт, который создал файл cookie, может его прочитать.
Произошла ошибка при настройке вашего пользовательского файла cookie
Произошла ошибка при настройке вашего пользовательского файла cookie
Этот сайт использует файлы cookie для повышения производительности. Если ваш браузер не принимает файлы cookie, вы не можете просматривать этот сайт.
Настройка вашего браузера для приема файлов cookie
Существует множество причин, по которым cookie не может быть установлен правильно. Ниже приведены наиболее частые причины:
- В вашем браузере отключены файлы cookie. Вам необходимо сбросить настройки вашего браузера, чтобы он принимал файлы cookie, или чтобы спросить вас, хотите ли вы принимать файлы cookie.
- Ваш браузер спрашивает вас, хотите ли вы принимать файлы cookie, и вы отказались.
Чтобы принять файлы cookie с этого сайта, используйте кнопку «Назад» и примите файлы cookie. - Ваш браузер не поддерживает файлы cookie. Если вы подозреваете это, попробуйте другой браузер.
- Дата на вашем компьютере в прошлом. Если часы вашего компьютера показывают дату до 1 января 1970 г.,
браузер автоматически забудет файл cookie. Чтобы исправить это, установите правильное время и дату на своем компьютере. - Вы установили приложение, которое отслеживает или блокирует установку файлов cookie. Вы должны отключить приложение при входе в систему или проконсультироваться с системным администратором.
 Вы должны отключить приложение при входе в систему или проконсультироваться с системным администратором.
Вы должны отключить приложение при входе в систему или проконсультироваться с системным администратором.Почему этому сайту требуются файлы cookie?
Этот сайт использует файлы cookie для повышения производительности, запоминая, что вы вошли в систему, когда переходите со страницы на страницу. Чтобы предоставить доступ без файлов cookie
потребует, чтобы сайт создавал новый сеанс для каждой посещаемой страницы, что замедляет работу системы до неприемлемого уровня.
Что сохраняется в файле cookie?
Этот сайт не хранит ничего, кроме автоматически сгенерированного идентификатора сеанса в cookie; никакая другая информация не фиксируется.
Как правило, в cookie-файлах может храниться только информация, которую вы предоставляете, или выбор, который вы делаете при посещении веб-сайта. Например, сайт
не может определить ваше имя электронной почты, пока вы не введете его. Разрешение веб-сайту создавать файлы cookie не дает этому или любому другому сайту доступа к
остальной части вашего компьютера, и только сайт, который создал файл cookie, может его прочитать.
Ключевая роль α-токсина в смертельной пневмонии, вызванной Staphylococcus aureus Тип последовательности 398
Редактору :
Пневмония — широко распространенное и тяжелое заболевание и ведущая причина инфекций, связанных с оказанием медицинской помощи. Золотистый стафилококк — важнейший возбудитель всех типов пневмонии (1). S. aureus тип последовательности (ST) 398 представляет собой клон, который традиционно ассоциируется с животноводством (2). Как метициллин-чувствительный (MSSA), так и метициллин-устойчивый ST398 преобладают у домашнего скота, но только ST398 MSSA, как сообщается, распространяется среди людей (3).Эти потенциально адаптированные к человеку штаммы ST398 MSSA в первую очередь связаны с инфекциями кожи и кровотока (4). Примечательно, что практически отсутствуют данные о роли факторов вирулентности в изолятах линии ST398, полученные с помощью передовых молекулярных исследований.
Мы провели проспективное исследование в больнице Медицинской школы Ботукату, Бразилия, с ноября 2011 года по август 2013 года, которое включало всех взрослых пациентов в отделении интенсивной терапии с ИВЛ (n = 270). S. aureus был изолирован от 47 пациентов, у 27 из которых развилась пневмония. Устойчивость к метициллину возникла у 22 изолятов (47%) и чаще встречалась при госпитальной пневмонии (7 из 9; 78%), чем при внебольничной пневмонии (1 из 7; 14%) или только при колонизации (без прогрессирования респираторной инфекции). , 6 из 18; 33%) изолятов.
S. aureus был изолирован от 47 пациентов, у 27 из которых развилась пневмония. Устойчивость к метициллину возникла у 22 изолятов (47%) и чаще встречалась при госпитальной пневмонии (7 из 9; 78%), чем при внебольничной пневмонии (1 из 7; 14%) или только при колонизации (без прогрессирования респираторной инфекции). , 6 из 18; 33%) изолятов.
Мультилокусное типирование последовательностей показало, что значительное количество изолятов (пять; 11%) принадлежало к линии ST398. Интересно, что частота случаев пневмонии была самой высокой среди линии ST398 (80%), как и уровень летальности в результате пневмонии (три из четырех случаев пневмонии; 75%).Три (27%) из 11 смертельных случаев пневмонии за отчетный период были вызваны изолятами ST398. Все изоляты ST398 были метициллин-чувствительными, положительными по генам chp и scn , которые, как было обнаружено, были связаны с адаптированным к человеку ST398 MSSA (3), и принадлежали к spa типу t1451. Краткие отчеты о трех случаях летальной пневмонии, ассоциированной с ST398, показаны в. Примечательно, что был, по крайней мере, один случай смертельной пневмонии ST398, при которой пациент не сообщил о контакте с животными, что свидетельствует о внутрибольничном заражении инфекционного штамма.
Краткие отчеты о трех случаях летальной пневмонии, ассоциированной с ST398, показаны в. Примечательно, что был, по крайней мере, один случай смертельной пневмонии ST398, при которой пациент не сообщил о контакте с животными, что свидетельствует о внутрибольничном заражении инфекционного штамма.
Таблица 1.
Краткие отчеты о случаях смертельной пневмонии, вызванных изолятами ST398
| Пациент | Изолят | Краткое описание случая |
|---|---|---|
| 39-летний мужчина | A16 | Больной поступил в реанимацию с травмой головы и ушибом легкого. У него развилась инфекция кожи и мягких тканей, которую лечили цефепимом и клиндамицином. Он умер от пневмонии, развившейся в течение 48 часов после госпитализации. Пациент был сельским рабочим. Пациент был сельским рабочим. |
| Мужчина 61 год | A19 | Пациент поступил в отделение интенсивной терапии с хронической почечной недостаточностью с острым обострением и необходимостью диализа. Он лечился амоксициллином и клавулановой кислотой из-за синусита в течение 3 дней после госпитализации. После этого лечение было изменено на имипенем и ванкомицин, поскольку у пациента развилась искусственная вентиляция легких. Пациент умер от вентилятор-ассоциированной пневмонии.Он сообщил, что контакта с животными не было. |
| Мужчина 76 лет | A52 | Больной поступил в реанимацию по поводу инфекции мочевыводящих путей с септическим шоком. Он получил имипенем, полимиксин E и линезолид. Он умер от пневмонии, связанной с госпиталем, не связанной с ИВЛ. Он сообщил о контактах со свиньями и домашней птицей. |
Мы не обнаружили значимой корреляции между наличием ключевых генов вирулентности и пневмонией или летальным исходом.Вдохновленные нашими недавними открытиями о том, что тяжесть стафилококковой инфекции кожи в значительной степени зависит от уровней экспрессии генов (5), мы предположили, что это также может иметь место в случае пневмонии ST398. Мы сосредоточили внимание на основных факторах, влияющих на стафилококковую инфекцию легких: α-токсин, фенолрастворимые пептиды модулина (PSM) и протеин A (6–8). Гены, кодирующие лейкоцидин Пантона-Валентайна, не присутствовали в наших изолятах ST398. Когда мы сравнивали все изоляты пневмонии, изоляты ST398 показали значительно более высокие уровни экспрессии in vitro и наиболее цитолитического PSM, PSMα3, δ-токсина (показание функциональности регулятора дополнительного гена Agr) и α-токсина, чем не -ST398 изоляты ().Различия в других PSM были аналогичными (данные не показаны). Кроме того, уровни PSM и α-токсина были подобны или превышали уровни, обнаруженные в высоковирулентном, ассоциированном с сообществом метициллин-устойчивом клоне S. aureus USA300, который, как известно, сильно экспрессирует PSM и α-токсин и потенциал вирулентности которого зависит от те цитолизины (6, 9) (). Примечательно, что уровни ST398 PSM и α-токсина сильно превышали уровни, обнаруженные в стандартном штамме ST398 S0385, метициллин-резистентном штамме S.aureus из случая эндокардита человека (10). Напротив, продукция отрицательно регулируемого Agr белка А была значительно ниже в изолятах ST398 по сравнению с другими изолятами (). Таким образом, наши данные свидетельствовали о высокофункциональной системе Agr в изолятах ST398, что мы подтвердили количественной полимеразной цепной реакцией РНКIII в реальном времени (). Кроме того, среди изолятов, вызвавших смертельную пневмонию, высокая продукция PSMα и α-токсина наблюдалась только у штаммов ST398, что указывает на особую роль пептидов PSMα и α-токсина в необычайной вирулентности изолятов ST398.Изоляты ST398 продемонстрировали выраженную способность лизировать эритроциты и нейтрофилы человека (), демонстрируя, что высокие уровни экспрессии цитолизина приводят к сильной цитолитической способности.
Молекулярное исследование фатальной пневмонии, вызванной типом последовательности (ST) 398. ( A ) In vitro Уровни продукции фенолрастворимого модулялина (PSM) α3 и δ-токсина (по данным высокоэффективной жидкостной хроматографии / масс-спектрометрии ), а также α-токсин и белок А (по данным вестерн-блот-денситометрии) всех изолятов пневмонии (здесь и во всех других экспериментах: выращивание до стационарной фазы роста в триптическом соевом бульоне).Значения, соответствующие смертельным случаям, показаны красным цветом . **** P <0,0001 (непарный тест t ). ( B и C ) Сравнение уровней продукции in vitro и PSM ( B ) и α-токсина ( C ) с уровнями USA300 (изолят USA300 SF8300 использовался для всех анализов в настоящее время. исследование) и изолята эндокардита ST398 S0385. **** P <0,0001 (по сравнению с USA300 и S0385, односторонний дисперсионный анализ [ANOVA]).( D ) Уровни экспрессии РНКIII с помощью количественной полимеразной цепной реакции в реальном времени. * P <0,05; ** P <0,01; *** P <0,001 (односторонний дисперсионный анализ; черные звездочки, по сравнению с USA300; серые звездочки, по сравнению с S0385). ( E ) Лизис нейтрофилов. Фильтраты культур измеряли на их способность лизировать нейтрофилы человека путем высвобождения лактатдегидрогеназы. Три столбца для каждого штамма представляют собой уменьшающиеся разведения: 1:10; 1: 5; и 1: 2.( F ) Лизис эритроцитов человека. Фильтраты культур использовали в данных разведениях. ( G – I ) Модель пневмонии мыши. Мышей интраназально инфицировали 1 × 10 8 колониеобразующих единиц, и в течение всего 72 часов наблюдали за развитием болезни. (G) Сравнение изолятов пневмонии ST398 с USA300 и S0385. n = 25 мышей на группу. **** P <0,0001 (лог-ранговый тест по сравнению с данными, полученными с помощью S0385). ( H ) Гистология. Микроскопические изображения легочной ткани; увеличение 40 ×. Белые стрелки указывают на бактериальные колонии, черные стрелки указывают на некротические сосуды. Показано изображение легочной ткани мыши, инфицированной штаммом A16; Патология в легких мышей, инфицированных другими штаммами ST398 и USA300, была сходной. ( I ) Сравнение изолята ST398 A19 дикого типа с изогенными штаммами с делецией hla и psm α. n = 10 мышей на группу. * P <0,05 (лог-ранговый тест; по сравнению с данными, полученными для штамма A19 дикого типа).( A – F ) Планки ошибок показывают ± SD.
В модели пневмонии у мышей () уровень смертности мышей, инфицированных тремя штаммами ST398, был высоким, по крайней мере равным таковому, вызванному штаммом USA300, и значительно выше, чем у мышей, вызванных штаммом S0385. Соответственно, гистологический анализ продемонстрировал обширную патологию в легких мышей, инфицированных штаммами ST398 или USA300, которые все проявляли признаки тяжелой и мультифокальной некротической пневмонии с острым воспалением, некротическим васкулитом, тромбозом и большим количеством бактериальных колоний в определенной степени. это было неотличимо между группами (показано для штамма A16).У мышей, инфицированных штаммом S0385, напротив, наблюдались только признаки умеренной перибронхиолярной пневмонии и отсутствие бактериальных колоний (). Таким образом, модель пневмонии на мышах хорошо отражала клинические результаты и подтвердила высокую вирулентность изолятов ST398.
Затем, чтобы проанализировать влияние PSMα и α-токсина, мы получили мутанты с делецией изогенного гена в опероне psm α и гене hla в изоляте A19. Оба мутанта psm α и hla вызвали меньшее количество смертей, чем изолят A19 дикого типа ().Однако только показатели выживаемости мутанта hla значительно отличались от таковых у штамма дикого типа, при этом ни одно животное, инфицированное мутантом A19 hla , не погибло от инфекции. Эти результаты приписывают центральную функцию α-токсину и высокой экспрессии α-токсина в патогенезе фатальной пневмонии, вызванной ST398 S. aureus .
В заключение, наше исследование идентифицирует ST398 MSSA как опасный и очень вирулентный возникающий источник фатальной пневмонии. Более того, хотя наши результаты подчеркивают зависимость от штаммов, они согласуются с представлением о решающей роли альфа-токсина в легочной инфекции.Наконец, наши результаты требуют мер наблюдения, анализирующих не только устойчивость к антибиотикам, но и потенциал вирулентности S. aureus как важный фактор, влияющий на исход инфекции.
Отключение длинной некодирующей РНК KCNQ1OT1 облегчает LPS-индуцированное повреждение легких, регулируя ось miR-370-3p / FOXM1 при детской пневмонии | BMC Pulmonary Medicine
Пациенты и сбор крови
Всего 24 ребенка (12 мальчиков и 12 девочек; средний возраст ± стандартное отклонение, 12.4 ± 1,18 года; возрастной диапазон 8–14 лет) с пневмонией были включены в настоящее исследование. Одновременно в качестве контроля были выбраны 24 здоровых ребенка с одинаковым полом и возрастом (средний возраст ± стандартное отклонение 10,3 ± 2,24 года; возрастной диапазон 7–14 лет). Диагностика пневмонии проводилась в соответствии с рекомендациями Всемирной организации здравоохранения по острым респираторным инфекциям [28]. Ни один из пациентов не получал никакого лечения до сбора крови, и пациенты с другими осложнениями были исключены.Периферическая венозная кровь (5 мл) была взята у всех участников. Это исследование было одобрено этическим комитетом Народной больницы Шоугуана (№ 20200905), и законные опекуны всех участников предоставили письменное информированное согласие.
Культивирование и обработка клеток
Нормальные диплоидные фибробласты легких человеческого эмбриона (клетки WI-38) были приобретены из Американской коллекции типовых культур (АТСС; Манассас, Вирджиния, США). Клетки WI-38 культивировали в среде Игла, модифицированной Дульбекко, содержащей 10% фетальной бычьей сыворотки, 100 мкг / мл стрептомицина и 100 Ед / мл пенициллина.Все клетки поддерживали при 37 ° C в увлажненной атмосфере, содержащей 5% CO 2 . Чтобы моделировать повреждение клеток, связанное с пневмонией, in vitro, клетки WI-38 стимулировали LPS (10 мкг / мл) в течение 12 часов [29], а клетки без обработки LPS служили контролем.
Трансфекция клеток
Малый интерферирующий (si) -KCNQ1OT1, si-негативный контроль (NC), миметики miR-370-3p, миметики-NC, ингибитор miR-370-3p, ингибитор-NC, pcDNA-FOXM1 и pcDNA -ЧПУ были приобретены в RiboBio (Гуанчжоу, Китай).Вышеуказанные факторы трансфицировали в клетки WI-38 с использованием Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) в течение 48 часов в соответствии с руководством пользователя.
Количественная полимеразная цепная реакция в реальном времени (qRT-PCR)
Реагент TRIzol (Invitrogen) использовали для выделения общей РНК из клеток WI-38 и образцов сыворотки. Набор реагентов PrimeScript RT (TaKaRa, Далянь, Китай) использовали для создания комплементарной ДНК (кДНК). qRT-PCR выполняли с использованием SYBR Green qPCR SuperMix (Invitrogen). Праймеры, приобретенные у TaKaRa, перечислены в таблице 1.Программа ПЦР-амплификации была следующей: 94 ° C в течение 10 минут, затем 40 циклов при 94 ° C в течение 15 секунд, 56 ° C в течение 30 секунд, 72 ° C в течение 1 минуты и 72 ° C в течение 10 минут. Относительные уровни экспрессии KCNQ1OT1, FOXM1 и miR-370-3p рассчитывали с использованием метода 2 — ΔΔCt . KCNQ1OT1 и FOXM1 были нормализованы до β-актина, а miR-370-3p нормализованы до U6.
Таблица 1 Праймеры для полимеразной цепной реакции в реальном времени (qRT-PCR) в настоящем исследовании
Анализ двойного люциферазного репортера (DLR)
Часть 3′-UTR KCNQ1OT1 или FOXM1, несущая потенциальные сайты связывания для miR-370- 3p вводили в вектор psiCHECK-2 (Promega, Мэдисон, Висконсин, США) для конструирования вектора wt KCNQ1OT1 или вектора FOXM1 wt.Аналогично, 3′-UTR-фрагмент KCNQ1OT1 или FOXM1, включая мутированные сайты связывания в комплементарных последовательностях для miR-370-3p, был клонирован в вектор psiCHECK-2 (Promega) для создания мутного вектора KCNQ1OT1 или мутного вектора FOXM1. Впоследствии клетки WI-38 котрансфицировали одним из вышеуказанных векторов вместе с имитаторами miR-370-3p или имитаторами-NC с использованием липофектамина 3000 (Invitrogen). Относительную активность люциферазы через 24 часа после трансфекции определяли с использованием системы анализа репортера люциферазы (Promega).
Анализ иммунопреципитации (RIP) связывающего РНК белка
Анализ RIP выполняли с использованием набора EZ-Magna RIP (Millipore, Billerica, MA, USA). Вкратце, клетки WI-38 трансфицировали миметиками-NC или miR-370-3p в течение 48 ч, а затем лизировали в полном буфере для лизиса RIP. Клеточные экстракты (100 мкл) инкубировали с буфером RIP, содержащим магнитные шарики, конъюгированные с человеческим антителом против Ago2 (основной белок РНК-индуцированного комплекса сайленсинга, который связывается с миРНК и целевыми мРНК) (Abcam, Кембридж, Массачусетс, США) или IgG мыши (Abcam) (контроль).Иммунопреципитированные РНК очищали и использовали для qRT-PCR для обнаружения экспрессии FOXM1.
Анализ 3- (4,5-диметил-2-тиазолил) -2,5-дифенил-2-H-тетразолийбромида (МТТ)
Клетки WI-38 высевали в 96-луночные планшеты (5 × 10 3 клеток / лунку). После инкубации в течение 48 часов в каждую лунку добавляли 20 мкл МТТ (Sigma-Aldrich, Сент-Луис, Миссури, США) и инкубировали при 37 ° C в течение 4 часов. Затем добавляли диметилсульфоксид (150 мкл) и перемешивали в течение 10 мин. Оптическую плотность каждой лунки при 490 нм измеряли с помощью считывающего устройства для микропланшетов (Bio-Rad, Hercules, Калифорния, США).
Оценка апоптоза клеток
Апоптоз клеток оценивали с использованием набора аннексина V и пропидия иодида (PI), конъюгированного с флуоресцеинизотиоцианатом (FITC) (BD Biosciences, Сан-Диего, Калифорния, США). Вкратце, клетки WI-38 промывали фосфатно-солевым буфером и окрашивали аннексином V-FITC (5 мкл) и PI (5 мкл) в течение 30 минут при 25 ° C в темноте. Проточная цитометрия использовалась для тестирования апоптотических клеток, и данные были проанализированы с использованием программного обеспечения FlowJo (Tree Star Inc., Ashland, OR, США).
Вестерн-блоттинг
Белки из клеток WI-38 экстрагировали с использованием буфера для лизиса RIPA (Beyotime, Шанхай, Китай).Белки разделяли с помощью гелей для электрофореза в полиакриламидном геле додецилсульфата натрия и переносили на поливинилиденфторидные мембраны. После блокирования 5% бычьим сывороточным альбумином в течение 2 ч мембраны инкубировали с первичными антителами против FOXM1 (1: 1000, ab207298, Abcam, Кембридж, Массачусетс, США) и α-тубулином (1: 2000, ab52866, Abcam) при 4 ° C в течение ночи. Затем мембраны промывали трис-буферным физиологическим раствором с Tween 20 и добавляли вторичное антитело (1: 2000; ab205718, Abcam) для культивирования в течение 1 часа.Наконец, полосы белков визуализировали с помощью Bio-Rad Gel Doc EZ Imager (Bio-Rad). Относительную экспрессию белка FOXM1, нормализованную по α-тубулину, количественно оценивали с использованием системы ChemiDoc XRS (Bio-Rad).
Создание мышиной модели LPS-индуцированного повреждения легких
Самцов мышей BALB / c (без специфических патогенов, шестинедельный возраст, вес 17-19 г) были приобретены в Esebio (Шанхай, Китай). Все мыши содержались в условиях постоянной температуры и влажности и имели свободный доступ к пище и воде.Чтобы вызвать связанное с пневмонией повреждение легких in vivo, мышей анестезировали внутрибрюшинной инъекцией 50 мг / кг пентобарбитала натрия, затем интратрахеально вводили 5 мг / кг ЛПС (Сент-Луис, Миссури, США) (растворенный в 50 мкл физиологического раствора) [30 ]. Мышам контрольной группы вводили 50 мкл физиологического раствора. Эксперименты на животных проводили после получения одобрения этического комитета Народной больницы Шоугуана (№ 20200905) в соответствии с Руководством по уходу и использованию лабораторных животных.
Обработки и сбор образцов
Рекомбинантные аденовирусы, содержащие короткую шпильочную РНК KCNQ1OT1 (Ad-sh-KCNQ1OT1), FOXM1 (Ad-FOXM1), пустой аденовирус NC (Ad-NC), antagomiR-370-3p и antagomiR-NC были куплены в Ribobio (Гуанчжоу, Китай). Вышеуказанные аденовирусы (100 мкл, 1 × 10 8 БОЕ / мл) и антагомиры (50 мг / кг массы тела) вводили мышам внутривенно за два дня до индукции модели. После обработки LPS в течение трех дней всех мышей анестезировали внутрибрюшинной инъекцией 50 мг / кг пентобарбитала натрия.Жидкость бронхоальвеолярного лаважа (ЖБАЛ) (1,4 мл) собирали для иммуноферментного анализа (ELISA). Затем мышей умерщвляли путем смещения шейных позвонков и вырезали правое легкое, чтобы измерить соотношение влажное / сухое легкое. Левое легкое использовалось для окрашивания гематоксилин-эозином (HE).
ELISA
Уровни фактора некроза опухоли-α (TNF-α), интерлейкина-6 (IL-6) и IL-1β в супернатантах культур клеток WI-38 и мышей BALB / c измеряли с использованием специфических Наборы ELISA (R&D Systems China, Шанхай, Китай).Считывающее устройство для микропланшетов (Molecular Devices, Саннивейл, Калифорния, США) использовали для исследования оптической плотности при 450 нм.
Окрашивание HE
Ткани правого легкого фиксировали в 4% параформальдегиде в течение 24 часов, дегидратировали в сериях градуированных этанолов, заливали парафином и разрезали на пять срезов. После окрашивания HE наблюдали патологические изменения в тканях легких под световым микроскопом (Olympus, Токио, Япония). Оценка травм рассчитывалась следующим образом: 0 — нет повреждений; l, легкое повреждение; 2, средний урон; 3, серьезное повреждение; и 4, серьезное повреждение [31].
Статистический анализ
Все эксперименты проводились независимо три раза. Для статистического анализа использовали SPSS версии 21.0 (IBM Software, New York, NY, USA). Данные в этом исследовании отображаются как среднее значение ± стандартное отклонение. Различия между двумя группами анализировали с помощью t-критерия Стьюдента. Для сравнения между несколькими группами применяли односторонний дисперсионный анализ с апостериорным тестом Тьюки. P <0,05 указывает на статистическую значимость.
Произошла ошибка при настройке пользовательского файла cookie
Этот сайт использует файлы cookie для повышения производительности.Если ваш браузер не принимает файлы cookie, вы не можете просматривать этот сайт.
Настройка вашего браузера для приема файлов cookie
Существует множество причин, по которым cookie не может быть установлен правильно. Ниже приведены наиболее частые причины:
- В вашем браузере отключены файлы cookie. Вам необходимо сбросить настройки вашего браузера, чтобы он принимал файлы cookie, или чтобы спросить вас, хотите ли вы принимать файлы cookie.
- Ваш браузер спрашивает вас, хотите ли вы принимать файлы cookie, и вы отказались.Чтобы принять файлы cookie с этого сайта, используйте кнопку «Назад» и примите файлы cookie.
- Ваш браузер не поддерживает файлы cookie. Если вы подозреваете это, попробуйте другой браузер.
- Дата на вашем компьютере в прошлом. Если часы вашего компьютера показывают дату до 1 января 1970 г.,
браузер автоматически забудет файл cookie. Чтобы исправить это, установите правильное время и дату на своем компьютере. - Вы установили приложение, которое отслеживает или блокирует установку файлов cookie.Вы должны отключить приложение при входе в систему или проконсультироваться с системным администратором.
Почему этому сайту требуются файлы cookie?
Этот сайт использует файлы cookie для повышения производительности, запоминая, что вы вошли в систему, когда переходите со страницы на страницу. Чтобы предоставить доступ без файлов cookie
потребует, чтобы сайт создавал новый сеанс для каждой посещаемой страницы, что замедляет работу системы до неприемлемого уровня.
Что сохраняется в файле cookie?
Этот сайт не хранит ничего, кроме автоматически сгенерированного идентификатора сеанса в cookie; никакая другая информация не фиксируется.
Как правило, в cookie-файлах может храниться только информация, которую вы предоставляете, или выбор, который вы делаете при посещении веб-сайта. Например, сайт
не может определить ваше имя электронной почты, пока вы не введете его. Разрешение веб-сайту создавать файлы cookie не дает этому или любому другому сайту доступа к
остальной части вашего компьютера, и только сайт, который создал файл cookie, может его прочитать.
% PDF-1.7
%
544 0 объект
>
эндобдж
xref
544 119
0000000016 00000 н.
0000003571 00000 н.
0000003761 00000 н.
0000003797 00000 н.
0000004503 00000 н.
0000004562 00000 н.
0000004700 00000 н.
0000004839 00000 н.
0000004978 00000 н.
0000005117 00000 н.
0000005256 00000 н.
0000005393 00000 п.
0000005525 00000 н.
0000006062 00000 н.
0000006174 00000 н.
0000006201 00000 н.
0000006608 00000 н.
0000006918 00000 н.
0000007032 00000 н.
0000007362 00000 н.
0000007611 00000 п.
0000008057 00000 н.
0000008783 00000 н.
0000008820 00000 н.
0000008847 00000 н.
0000012319 00000 п.
0000015817 00000 п.
0000019153 00000 п.
0000022015 00000 н.
0000022148 00000 п.
0000022287 00000 п.
0000022547 00000 п.
0000022981 00000 п.
0000023342 00000 п.
0000023369 00000 п.
0000023946 00000 п.
0000023973 00000 п.
0000024643 00000 п.
0000024952 00000 п.
0000028812 00000 п.
0000028944 00000 п.
0000029388 00000 п.
0000029782 00000 п.
0000029809 00000 п.
0000030469 00000 п.
0000030724 00000 п.
0000031214 00000 п.
0000034276 00000 п.
0000034633 00000 п.
0000034882 00000 п.
0000037611 00000 п.
0000040210 00000 п.
0000042860 00000 п.
0000042930 00000 н.
0000043015 00000 п.
0000046836 00000 п.
0000047098 00000 п.
0000047268 00000 п.
0000047338 00000 п.
0000047408 00000 п.
0000047911 00000 п.
0000047981 00000 п.
0000048115 00000 п.
0000070952 00000 п.
0000071227 00000 п.
0000071736 00000 п.
0000100905 00000 н.
0000119305 00000 н.
0000140265 00000 н.
0000140399 00000 н.
0000140469 00000 н.
0000140578 00000 н.
0000173584 00000 н.
0000173847 00000 н.
0000174382 00000 н.
0000174533 00000 н.
0000201770 00000 н.
0000228173 00000 н.
0000228442 00000 н.
0000247733 00000 н.
0000248458 00000 н.
0000248721 00000 н.
0000273648 00000 н.
0000273911 00000 н.
0000274396 00000 н.
0000290622 00000 н.
0000290878 00000 н.
0000291273 00000 н.
0000298223 00000 н.
0000298262 00000 н.
0000328975 00000 н.
0000329014 00000 н.
0000359727 00000 н.
0000359766 00000 н.
0000396455 00000 н.
0000396494 00000 н.
0000396882 00000 н.
0000396979 00000 п.
0000397168 00000 н.
0000397606 00000 н.
0000398025 00000 н.
0000398441 00000 н.
0000398799 00000 н.
0000399106 00000 н.
0000418165 00000 н.
0000418423 00000 н.
0000418824 00000 н.
0000419226 00000 п.
0000419655 00000 н.
0000420054 00000 н.
0000426219 00000 н.
0000426258 00000 н.
0000435994 00000 н.
0000436908 00000 н.
0000436996 00000 н.
0000437084 00000 н.
0000437167 00000 н.
0000437250 00000 н.
0000002676 00000 н.
трейлер
] / Назад 1016439 >>
startxref
0
%% EOF
662 0 объект
> поток
h ޤ TmHSQ ~ k ݽ m2ù9Xtnns ~ B.07u ڢ UT * «_? +) Ad`D (RR ݜ> py
Ключевые механизмы, управляющие разрешением воспаления легких
Острые воспалительные реакции инициируются травмой, инфекцией и раздражением, которые, в свою очередь, защищают хозяина от системной инфекции и помогают восстанавливают гомеостаз тканей [1]. Таким образом, воспаление представляет собой важный защитный механизм, который является защитным и жизненно важным для здоровья [2, 3]. Как правило, молекулярные события и клеточные взаимодействия, преобладающие во время острых воспалительных реакций, эффективны для минимизации надвигающегося повреждения, инфекции или раздражения. , что, что важно, приводит к восстановлению гомеостаза тканей и, таким образом, к полному разрешению острого воспалительного ответа.Однако если возникает острая воспалительная реакция, неконтролируемая по величине или продолжительности, она может привести к заболеванию [1, 3]. В легких нарушение регуляции острого воспаления может привести к повреждению легких, что способствует развитию легочного фиброза, который серьезно нарушает основные процессы газообмена. Следовательно, существует множество механизмов, которые жестко регулируют тяжесть и продолжительность воспаления легких. Если не решить проблему, острое повреждение легких (ОПЛ) и / или воспаление легких может перейти в хроническое воспаление, которое возникает при таких заболеваниях легких, как острый респираторный дистресс-синдром (ОРДС), астма, муковисцидоз (МВ) и хроническая обструктивная болезнь легких (ХОБЛ). [1].
Расслабление воспаления ранее рассматривалось как пассивный процесс с ограниченным пониманием механизмов, регулирующих разрешение воспаления. Однако на протяжении многих лет существенные исследования в этой области определили разрешение воспаления как активный и строго регулируемый клеточный и биохимический процесс. В настоящее время известно, что существует множество молекулярных медиаторов воспаления, включая многие про- и противовоспалительные цитокины и хемокины, с ослаблением эффектов провоспалительных медиаторов, способствующих успешному «выключению» воспаления [4].Совсем недавно было обнаружено несколько эндогенных про-рассасывающих биоактивных липидных медиаторов (иммунорезольвентов), таких как липоксины, резольвины, протектины и марезины, которые активно участвуют в «запрограммированном разрешении», которое успешно прекращает воспаление [5–8]. Другие ключевые процессы, управляющие успешным разрешением воспаления, включают фагоцитарный клиренс апоптотических клеток [9, 10] во время процесса, называемого эффероцитозом, который также приводит к фагоцитозным клеткам, переключая фенотип с провоспалительной клетки на более противовоспалительную. / про-разрешающий фенотип [10, 11].Также, относящийся к мукоцилиарному клиренсу легких инфекционные агенты, аллергены, инородные частицы и эффетивные клетки возникают [12]. Этот обзор охватывает клеточные механизмы и главные биохимические медиаторы, участвующие в разрешении воспаления легких и восстановлении поврежденных тканей, с особым акцентом на воспаление легких с преобладанием нейтрофилов / эозинофилов и фармакологические подходы к разрешению болезни [13–16].
Клетки врожденной иммунной системы
Антиген-независимый врожденный иммунитет обеспечивает первую линию лейкоцитарной защиты от вторжения микроорганизмов, во время которой воспаление является ранней ключевой реакцией на инфекцию, травму или раздражение.Врожденная иммунная защита при воспалении легких включает несколько типов клеток и их взаимодействие. К ним относятся лейкоциты, такие как полиморфноядерные гранулоциты (нейтрофилы, эозинофилы, базофилы) и агранулоциты (моноциты, макрофаги), эпителиальные / эндотелиальные клетки легких, тучные клетки, естественные киллеры (NK-клетки) и дендритные клетки. Эти клетки могут влиять на функцию других типов клеток, таких как врожденные лимфоидные клетки [17] и лимфоциты [18], которые конкретно не рассматриваются в данном обзоре.
Нейтрофилы
Нейтрофилы, составляющие 70% циркулирующих лейкоцитов крови человека, недолговечны в кровотоке, сохраняясь до 7-10 часов (хотя точный период времени в кровотоке остается спорным; см. Tak et al [ 19]). Однако во время воспалительного процесса или в ответ на химические раздражители они могут прожить до 48 часов или более. Эти клетки, обычно диаметром 12-15 мкм, содержат отчетливое многодольчатое ядро и четыре разных типа гранул: первичные (азурофильные), вторичные (специфические), желатиназные и секреторные.Эти гранулы содержат> 300 белков, которые участвуют в нескольких клеточных процессах, включая миграцию, адгезию и антимикробную активность [20]. Нейтрофилы очень универсальны и при воспалительном поражении быстро мигрируют в очаги травмы / инфекции, где они часто оказываются первыми и помогают защитить хозяина посредством фагоцитоза, дегрануляции, генерации активных форм кислорода (АФК) или высвобождения паутины хроматин посредством образования внеклеточных ловушек нейтрофилов (NET) для улавливания и уничтожения микроорганизмов.Кроме того, есть доказательства того, что нейтрофилы могут меняться с провоспалительного на противовоспалительный фенотип после успешного воспалительного ответа. В таком случае нейтрофилы перестают продуцировать и высвобождать провоспалительные медиаторы, такие как лейкотриен B 4 (LTB 4 ) и фактор активации тромбоцитов (PAF), и начинают продуцировать и высвобождать провоспалительные медиаторы, такие как провоспалительные биоактивные липиды (например, липоксины), которые усиливают фазу разрешения воспаления [21], для тщательного анализа свойств нейтрофилов, способствующих разрешению, [см. 22].Как только нейтрофилы выполнили свою физиологическую функцию, они обычно подвергаются апоптозу, который сохраняет целостность мембраны, предотвращая тем самым неконтролируемое высвобождение вредного клеточного содержимого и интернализованных микробов в непосредственную внеклеточную среду [23]. Специфичные для апоптоза клеточные изменения способствуют распознаванию и поглощению нейтрофилов фагоцитами, такими как макрофаги, дендритные клетки и эпителиальные клетки. Для успешного разрешения воспаления важно, чтобы нейтрофилы «выключались», подвергались апоптозу и успешно очищались.Если не разрешить острое воспаление, оно может привести к хроническому воспалению, при котором массовый приток нейтрофилов к локализованным окрестностям приводит к повреждению тканей хозяина. Это может происходить, когда избыток нейтрофилов продуцирует свободные радикалы во время генерации ROS, высвобождает протеолитическое и антимикробное содержимое гранул во время дегрануляции и экстернализации хроматина, усеянного ядерными, гранулярными и цитозольными белками с высокими антимикробными свойствами во время генерации NET. Более того, в условиях гипоксии (1% кислорода), которые обычно наблюдаются на протяжении всего воспаления, наблюдается выживание нейтрофилов, вызванное гипоксией, опосредованное гипоксией-индуцируемым фактором (HIF) -1α-зависимым ядерным фактором, усилителем легкой цепи каппа активированных В-клеток (NF). -ƘB) активность и экспрессия пролилгидроксилазы 3.Как лимфоциты и макрофаги, данные свидетельствуют о том, что нейтрофилы также могут существовать как гетерогенная популяция, проявляющая разные фенотипы [24, 25]. Интересно, что накопление последних данных указывает на то, что нейтрофилы, в зависимости от воспалительной реакции, могут быть способны покидать окрестности воспалительного сайта в результате процесса, называемого обратной миграцией [26–32]. Воспаление с преобладанием нейтрофилов связано с рядом воспалительных заболеваний легких, включая ALI / ARDS, CF, COPD, идиопатический фиброз легких (IPF), бронхоэктазы, атопическую / неатопическую астму и тяжелую астму, во время которой повышается количество нейтрофилов (нейтрофильная астма ).
Эозинофилы
Эти клетки составляют <5% лейкоцитов циркулирующей крови человека и могут выжить до 12 часов, но при необходимости обладают способностью продлевать свою жизнь как минимум на неделю. Они немного крупнее нейтрофилов диаметром 12-17 мкм, обладают двудольным ядром и легко идентифицируются при окрашивании по Романовскому (метиленовый синий и эозин). Подобно нейтрофилам, эозинофилы наполнены гранулами в своей цитоплазме; однако эозинофильные гранулы содержат различные компоненты, такие как эозинофильный катионный белок, основной основной белок, пероксидазу эозинофилов и нейротоксин, полученный из эозинофилов, которые цитотоксичны для эпителиальных клеток дыхательных путей.По прибытии к месту травмы / инфекции (особенно паразитарной инфекции) эозинофилы подвергаются дегрануляции, которая помогает устранить воспалительные стимулы, предотвращая дальнейшее воспаление. Эозинофилы также могут вносить вклад в защиту хозяина посредством высвобождения внеклеточных ловушек эозинофилов (EET), состоящих либо из митохондриальной, либо из ядерной ДНК [33, 34]. Эти EET вносят вклад в антимикробную защиту путем высвобождения митохондриальной ДНК, которая ассоциируется с гранулированными белками, происходящими из эозинофилов, способными захватывать и убивать микроорганизмы in vitro и in vivo [33].Эозинофилы также могут подвергаться апоптозу, который затем очищается посредством фагоцитоза макрофагами, модулируемыми IL-5 [35]. Эозинофилы способны проявлять как провоспалительный, так и противовоспалительный фенотип и функцию. Противовоспалительная активность эозинофилов включает важную регулирующую роль во время реакций гиперчувствительности за счет опосредованной эозинофильной пероксидазой инактивации LTB 4 , C 4 и D 4 [36]. Однако пероксидаза эозинофилов может также проявлять провоспалительную активность в областях воспаления, где присутствуют и тучные клетки, и эозинофилы, в основном за счет внеклеточного образования активных комплексов, образованных между пероксидазой эозинофилов и гранулами тучных клеток [37].Антивирусная активность также была документально подтверждена для человеческих эозинофилов и связанных с ними рибонуклеаз против респираторно-синцитиального вируса (in vitro) и для эозинофилов мышей и ассоциированных рибонуклеаз против вируса пневмонии мышей in vivo (для обзора см. Rosenberg and Domachowske [38]). Кроме того, эозинофилы человека и мыши способны проявлять противовирусную активность против парагриппа 1 в легких (распространенный респираторный вирус) [39]. Эозинофилы доминируют при аллергическом воспалении дыхательных путей [40], включая атопическую / неатопическую астму и аллергический ринит, и, как известно, участвуют в поддержании и восстановлении гомеостаза легких.
Базофилы
Базофилы представляют собой самые редкие из циркулирующих гранулоцитов человека, и их гранулы содержат множество веществ, включая гистамин, гепарин, серотонин, нейтральные протеазы и гидролазы. При стимуляции они способны высвобождать содержимое своих гранул и синтезировать медиаторы, включая биоактивные липиды и цитокины. Таким образом, при воздействии аллергенов они активируются и быстро дегранулируют, что усиливает воспаление при атопической / неатопической астме и аллергическом рините [41].
Моноциты / макрофаги
Моноциты содержат множество гранул меньшего размера, чем их гранулоцитарные аналоги, которые в основном содержат лизосомальные ферменты, которые способствуют разрушению интернализованных фагоцитированных микроорганизмов. При отсутствии воспаления моноциты обычно ограничиваются костным мозгом и кровью. Однако при воспалительном поражении они быстро мигрируют в воспаленную ткань и дифференцируются в крупные тканевые фагоцитарные макрофаги. В зависимости от микросреды макрофаги могут менять свой статус на различные фенотипы.Обратите внимание, что для удобства макрофаги были разделены на разные фенотипы; эта номенклатура неточна, и авторы отмечают, что макрофаги пластичны и могут меняться в зависимости от окружающей среды, происхождения и статуса активации. Например, они могут иметь более провоспалительный фенотип (часто обозначаемый как M1 или классические макрофаги), противовоспалительный фенотип (обозначаемый как M2 или альтернативные макрофаги) или про-разрешающий фенотип [11, 42, 43]. Переключение M1 может быть вызвано внутриклеточными патогенами, компонентами бактериальной клеточной стенки, такими как липополисахарид (LPS), липопротеинами и растворимыми медиаторами, такими как цитокины, интерферон гамма (IFNγ) и фактор некроза опухоли (TNF), которые, в свою очередь, приводят к высвобождению различные провоспалительные цитокины / медиаторы (IL-1, IL-6, IL-8, TNF, IFNγ, LTB 4 ), обостряющие воспаление, а также образование оксида азота (NO), которое способствует эффективному уничтожению микроорганизмов [44 ].Переключение M2 может быть вызвано различными паразитами и грибковыми клетками; иммунные комплексы; апоптотические клетки; и растворимые медиаторы, включая макрофагальный колониестимулирующий фактор (M-CSF), IL-4, IL-10, IL-13 и трансформирующий фактор роста бета (TGFβ) [45]. Макрофаги M2 блокируют высвобождение провоспалительных стимулов и высвобождают про-репаративные и противовоспалительные цитокины / медиаторы, такие как IL-10, TGFβ и простагландин E 2 (PGE 2 ). Более того, макрофаги M2 обладают повышенной фагоцитарной способностью, при этом их наиболее важной функцией является эффективный клиренс апоптозных клеток [46], что в значительной степени способствует успешному разрешению воспаления.Во время инфекции и повреждения легких миграция и удержание популяций моноцитов и макрофагов участвуют в запуске и поддержании воспаления легких [47].
Эпителиальные / эндотелиальные клетки легких
В дыхательных путях трахея в сочетании с главными бронхами составляет проксимальные хрящевые дыхательные пути и отвечает за проводимость вдыхаемого воздуха. Во время дыхания проксимальный псевдостратифицированный эпителий участвует в защите от токсинов окружающей среды и вторжения микроорганизмов.Напротив, более столбчатый эпителий расположен в дистальных отделах дыхательных путей, где расположено большое количество бокаловидных и мерцательных эпителиальных клеток. Бокаловидные эпителиальные клетки секретируют слизистую, которая обеспечивает смазку, помогая реснитчатым эпителиальным клеткам уносить микроорганизмы, частицы пыли и эффетирующие клетки от легких (мукоцилиарный клиренс). Микроваскулярный эндотелий легких и эпителиальная выстилка альвеол образуют основу газообменных воздухо-кровяных барьеров в легких. Этот барьер состоит из трех отдельных частей: крови, интерстиция и альвеолярного пространства [48].Альвеолярный эпителий состоит из альвеолярных клеток 1-го и 2-го типа (пневмоцитов), при этом газообмен осуществляется клетками 1-го типа, а выделение легочного сурфактанта осуществляется клетками 2-го типа, что поддерживает нормальную функцию легких за счет снижения поверхностного натяжения. . Альвеолярные макрофаги также являются наиболее многочисленными фагоцитами, присутствующими в альвеолярном пространстве легкого. Благодаря большой площади поверхности и постоянному натиску микроорганизмов и частиц, присутствующих в воздухе, легкие приобрели эффективные механизмы обнаружения микробов.У младенцев нарушения развития в обширном интерфейсе, где альвеолярные эндотелиальные клетки находятся прямо напротив альвеолярных эпителиальных клеток, могут привести к тяжелым респираторным осложнениям. В зрелых легких непоправимые изменения в структуре границы раздела кровь-газ способствуют фиброзным заболеваниям легких и эмфиземе легких, тогда как дисфункция барьера эндотелиальных / эпителиальных клеток легких является основным фактором ALI. Более того, при ALI / ARDS обширное повреждение барьеров эндотелиальных / эпителиальных клеток вызывает утечку отечной жидкости и воспалительных клеток в альвеолярные пространства, что приводит к гипоксемии и дыхательной недостаточности.Что касается репаративных возможностей, эндотелиальные клетки могут способствовать восстановлению эпителия в микроокружении легких [49].
Тучные клетки, NK-клетки и дендритные клетки
Считается, что тучные клетки участвуют в заживлении и заживлении ран и обнаруживаются в коже и слизистых / соединительных тканях, где в ответ на патогенный инсульт преимущественно концентрируются в слизистой / соединительной ткани для обеспечивают врожденную иммунную защиту. Тучные клетки (посредством дегрануляции) могут выделять мощные медиаторы воспаления, включая гистамины, протеазы, хемотаксические факторы, цитокины и метаболиты арахидоновой кислоты, которые воздействуют на сосудистую сеть, гладкие мышцы, соединительную ткань, слизистые железы и другие воспалительные клетки [50].Подобно эозинофилам и базофилам, тучные клетки участвуют в аллергическом воспалении дыхательных путей с повышенным количеством тучных клеток в легочных альвеолах и дыхательных путях, а также в астматических легких или в жидкости бронхиального альвеолярного лаважа (БАЛ) пациентов с IPF и саркоидозом [51]. Другие типы клеток, участвующие в воспалении легких, включают NK-клетки и дендритные клетки. NK-клетки представляют собой цитотоксические лимфоциты и могут играть как полезные, так и неблагоприятные роли при астме, ХОБЛ, гриппе и туберкулезе.Однако остается нехватка знаний о том, как функции этих клеток регулируются в уникальной тканевой среде каждого состояния [52]. Дендритные клетки являются посредниками между врожденной и адаптивной иммунной системами. В легких дендритные клетки создают межфазную клеточную поверхность между внешней средой и микросредой. Дендритные клетки легких играют важную роль в патогенезе астмы посредством регуляции гиперреактивности бронхов, рекрутирования эозинофилов / тучных клеток в места воспаления дыхательных путей и индукции гиперплазии бокаловидных клеток [53].
Смерть клеток
Процессы гибели клеток жестко регулируются, чтобы гарантировать успешное разрешение воспаления. Тем не менее, нарушение регуляции гибели клеток обычно препятствует процессу разрешения. В легких многочисленные процессы гибели клеток управляют воспалением. Понимание механизмов, регулирующих гибель клеток в легких, поможет идентифицировать новые терапевтические цели для ограничения / устранения воспаления и восстановления гомеостаза.
Апоптоз
Апоптоз гранулоцитов был предметом большого интереса в последние десятилетия, и есть убедительные доказательства того, что неспособность разрешить воспаление способствует многочисленным хроническим воспалительным состояниям и их манипуляциям, что, следовательно, предлагает новые потенциальные терапевтические цели.Воспалительные клетки обладают потенциалом воспламенения в тканевой среде хозяина и, при отсутствии соответствующей воспалительной «угрозы», могут вызывать повреждение ткани хозяина, вторичное по отношению к высвобождению гистотоксических медиаторов, таких как протеазы и активные формы кислорода. Возможно, одним из наиболее важных механизмов разрешения и восстановления гомеостаза тканей после острого воспалительного инсульта является способность накопленных мигрирующих гранулоцитов подвергаться иммунологически безмолвной запрограммированной гибели клеток, а именно апоптозу.Этот строго регулируемый, энергозависимый и сложный процесс включает скоординированное разрушение и упаковку содержимого воспалительных клеток для фагоцитарной очистки таким образом, который не вызывает иммунного ответа хозяина, способствует заживлению, а также способствует и поддерживает самотолерантность адаптивной иммунной системой. для создания иммунологической памяти. Кроме того, эта аккуратная упаковка содержимого клеток предотвращает утечку провоспалительных медиаторов и содержит гистотоксическое оружие, включая протеазы, продукцию активных форм кислорода и лизоцимы.Апоптоз гранулоцитов — это каспазозависимый процесс, который происходит после активации одного из двух основных путей, внутреннего и внешнего [1, 54]. Становится все более очевидным, что взаимная исключительность этих путей не так однозначна, как предполагалось ранее, и существует определенная степень перекрестного взаимодействия между молекулярным компонентом их выполнения, причем оба в конечном итоге зависят от действий каспаз по инициированию. клеточное самоубийство. Каспазы, семейство цистеин-аспарагиновых протеаз, являются критическими внутриклеточными медиаторами апоптоза, а также участвуют в воспалении и некрозе, таким образом предлагая многообещающую мишень для фармакологических манипуляций [55, 56].
Внутренний, или митохондриальный, путь возникает, когда баланс про- и антиапоптотических медиаторов белков семейства Bcl-2 склоняется в пользу гибели клеток, которая происходит в ответ на повреждение ДНК или стресс эндоплазматического ретикулума. В зрелом гранулоците проапоптотические члены семейства Bax, Bad, Bak и Bid подавляются их антиапоптотическими аналогами, Mcl-1, Bcl-xl и A1, таким образом поддерживая жизнеспособность клеток. В присутствии достаточного клеточного стресса они обходят это подавление и перемещаются из цитоплазмы в митохондрии, запуская развитие проницаемости митохондриальной внешней мембраны (MOMP).MOMP позволяет митохондриальным молекулам цитохрома C, Smac / DIABLO, Omi / HtrA2 и сериновым протеазам проникать в цитозоль, где цитохром C взаимодействует с Apaf-1 с образованием апоптосомы, которая в конечном итоге ответственна за гибель клеток через активацию прокаспазы 9. Образовавшаяся каспаза 9 вызывает расщепление «исполнителя» — каспазы 3, что приводит к фрагментации ДНК, перекрестному связыванию и деградации внутриклеточных белков и переключению мембранных рецепторов. Напротив, апоптоз, продвигающийся по внешнему пути «рецептора смерти», происходит в ответ на стимуляцию внеклеточными медиаторами, в первую очередь TNF, лигандом Fas и лигандом, индуцирующим апоптоз, связанным с фактором некроза опухоли альфа (TRAIL) [57].Эти межклеточные мессенджеры активируют рецепторы на плазматических мембранах гранулоцитов, в частности рецептор TRAIL (TRAIL-R), рецептор TNF 1 (TNFR1) и рецептор Fas (FasR), которые после связывания с их соответствующим лигандом стремятся объединиться. Сборки мембранных белков связываются с внутренними адаптерами, образуя белки домена смерти, которые привлекают кластеры цитозольной прокаспазы 8. Взаимодействие этих белков запускает внутриклеточный каскад, а именно, вызывающий смерть сигнальный комплекс (DISC), который завершается автокаталитическим расщеплением прокаспаза 8, которая затем снова приводит к апоптозу клетки через расщепление каспазы 3.Каспаза 8, генерируемая в ответ на внеклеточный лиганд Fas, является основным исполнителем перекрестного взаимодействия между внутренними и внешними путями, поскольку ее высвобождение запускает MOMP через расщепление Bid [58, 59].
После активации каспазы ядерная ДНК образует нуклеосомы, плотные пакеты генетического материала. Одновременно происходит изменение рецепторного профиля плазматической мембраны. Набор молекул, способствующих выживанию, который включает CD47 и CD31, заменен средой сигналов «найди меня» и «съешь меня», которые запускают распознавание и стимулируют поглощение умирающей клетки макрофагами или другими клетками с фагоцитарной способностью [ 10, 60].Сигналы «найди меня» испускаются апоптотическими клетками, которые впоследствии привлекают близлежащие фагоциты. У млекопитающих было идентифицировано несколько сигналов find-me, включая фракталкин (CX3CL1), лизофосфатидихолин (липидный медиатор), сфингозин-1-фосфат и нуклеотиды, включая аденозинтрифосфат и уридин-5′-трифосфат [61–64]. Сигналы Eat-me позволяют специфически распознавать апоптотические клетки через различные изменения клеточной поверхности, которые включают воздействие фосфатидилсерина (PS) на листок наружной мембраны, изменение эпитопа молекулы-1 внутриклеточной адгезии (ICAM1), воздействие кальретикулина и изменение клеточной поверхности. конфигурации заряда и гликозилирования (для обзора см. Gardai et al.[65]). Лучше всего описанный и наиболее эволюционно консервативный из этих сигналов eat-me — это экстернализация PS на листок внешней мембраны [66, 67], который вместе с ICAM3 и аннексином 1 способствует фагоцитозу. Кроме того, сигналы find-me, такие как нуклеотиды, фракталкин и липидные медиаторы, привлекают не только профессиональные фагоциты, но также могут способствовать захвату соседними клетками и другими непрофессиональными фагоцитами, включая клетки бронхиального эпителия [10, 68, 69]. В дополнение к предотвращению прямого, хотя и непреднамеренного повреждения тканей хозяина, этот механизм удаления ослабляет иммунный ответ и способствует разрешению, позволяя возобновить нормальный гомеостаз тканей.Апоптоз является важным механизмом выведения эффек- тивных клеток и успешного разрешения воспаления легких. Было показано, что апоптоз гранулоцитов задерживается при заболевании легких, а специфическая индукция апоптоза гранулоцитов может улучшить разрешение воспаления легких (подробные обзоры см. В [1, 14]).
Другие процессы гибели клеток в легких
В противоположность своему хорошо сдержанному аналогу (апоптозу) некроз приводит к потере целостности мембраны и неограниченному высвобождению внутриклеточного содержимого после травмы клетки.Высвобождение молекулярных паттернов, связанных с токсическим повреждением (DAMP), во внеклеточную среду, как правило, приводит к острой воспалительной реакции с притоком воспалительных клеток, паракринным эффектам на окружающие клетки с высвобождением провоспалительных медиаторов и значительным потенциалом разрушения тканей хозяина [70, 71]. Различные повреждения, включая инфекцию, химические вещества, физические травмы и дефицит питания, вызывают прямую потерю целостности мембран — так называемый первичный некроз.В ситуациях, когда не удается своевременно и в достаточной степени очищать апоптотические клетки фагоцитами, возникает вторичный некроз из-за неизбежного распада апоптотической клеточной мембраны, что может привести к поздней фазе воспалительной реакции, природа которой все еще остается обсуждается [10, 60, 68]. Растет признание важности DAMP в распространении острого воспалительного ответа в легких через взаимодействие с рецепторами распознавания патогенов (PRR) [72].Их активация способствует воспалению за счет транскрипции провоспалительных цитокинов и усиливает антимикробный ответ. Появляется все больше доказательств того, что вторичный некроз, некротическая судьба клетки после отказа фагоцитоза после того, как произошел апоптоз, способствует стойкому воспалению в ряде хронических состояний и, безусловно, очевидно, что некроз в контексте гиперактивной острой реакции может приводят к значительным долгосрочным последствиям в результате повреждения тканей и аберрантного ремоделирования.
В 2004 году был открыт новый процесс гибели клеток, который четко отделен от некроза и апоптоза, когда было обнаружено, что нейтрофилы человека могут генерировать NET [73] для врожденной иммунной защиты. NET состоят из деконденсированного ядерного хроматина, который контролируемым образом выводится во внеклеточную среду. Кроме того, нейтрофилы могут выделять митохондриальную ДНК [74]; однако митохондриальная ДНК в 100 000 раз менее распространена в NET, чем ядерная ДНК [75]. NET характеризуются тем, что ядерная мембрана полностью фрагментирована, при этом большая часть гранул растворяется, что обеспечивает прямой контакт и смешивание ядерных, цитоплазматических и гранулярных компонентов [76].На основе ДНК NETs расположены ядерные, гранулярные и цитозольные белки. Ядерные белки включают цитруллинированные гистоны и антимикробные пептиды (AMP), такие как кателицидин, LL37; азурофильные (первичные) белки гранул, такие как нейтрофильная эластаза (NE), катеспин G, миелопероксидаза (MPO) и α-дефенсины; специфические белки из вторичных и третичных гранул, такие как лактоферрин и желатиназа соответственно; или цитозольные белки, такие как комплекс цитозольных белков, кальпротектин [73, 77].Основные гистоны h3A, h3B, h4 и h5 составляют 70% всех NET-ассоциированных белков [77]. Гиперцитруллинирование гистонов, которое опосредует деконденсацию хроматина во время образования NET, опосредуется пептидиларгининдезиминазой 4 (PAD4) [78].
Образование NETs зависит от генерации ROS посредством активации никотинамидадениндинуклеотидфосфат (NADPH) оксидазы, полимеризации актиновых филаментов, а также от необходимости активации путей протеинкиназы C (PKC) перед НАДФН-оксидазой [76, 79, 80].NET обладают способностью улавливать и убивать как грамположительные, так и отрицательные бактерии, вирусы, грибы и более крупных паразитов [73, 81–83]; однако в настоящее время широко считается, что NET более эффективны для улавливания микроорганизмов, чем для уничтожения. При этом бактерии действительно обладают способностью ускользать и разрушать NET посредством множества механизмов, например, образования полисахаридов, которое вызывает электрохимическое отталкивание AMPs, или генерации ДНКазы, способствующей деградации хроматина [84, 85]. Этот процесс был впервые назван НЕТозом [86], поскольку считалось, что он присущ только нейтрофилам.Однако этот путь гибели клеток теперь обычно называют ЭТозом [87], и его можно обнаружить в ряде различных типов иммунных клеток, а также в гемоцитах низших беспозвоночных [88]. Раннее происхождение ETosis помогает объяснить некоторые из его патологических эффектов у млекопитающих, где ETosis можно рассматривать как палку о двух концах.
ETosis вовлечен в ряд хронических воспалительных заболеваний легких, включая ALI и ARDS, гриппозную пневмонию, муковисцидоз, астму, ХОБЛ и туберкулез.Отличительным признаком инфекции, связанной с ALI / ARDS и стерильным повреждением, является активация и последующая массовая миграция нейтрофилов в альвеолярное пространство, которая инициируется хемокинами, высвобождаемыми из макрофагов, нейтрофилов и эпителиальных клеток [89]. Активация нейтрофилов и образование NET в альвеолярном пространстве инициируются сильно локализованной концентрацией стимулирующих факторов. Повреждение альвеолярных эпителиальных клеток увеличивает проницаемость барьера между альвеолярным пространством и кровеносными сосудами, что также способствует высвобождению провоспалительным IL-8 эпителием.Это может привести к утечке отечной жидкости, содержащей большое количество проникающих нейтрофилов, в альвеолярное пространство. В альвеолах NET высвобождаются в ответ на производные от хозяина факторы, такие как гранулоцитарный / макрофагальный колониестимулирующий фактор (GM-CSF), фактор комплемента 5a (C5a), активированные тромбоциты и синглетный кислород. Затем NET вызывают вторичное повреждение эпителиальных клеток за счет высвобождения белков NET и генерации АФК, что приводит к хроническому воспалению. Мощные факторы повреждения легких, выделяемые NET, включают NE, который расщепляет эндотелиальный цитоскелет, а также E-cadherin и VE-cadherin, которые увеличивают проницаемость альвеолярно-капиллярного барьера [90].Другие производные NET компоненты, такие как cathespin G, могут разрушать противовоспалительные белки посредством продукции провоспалительных белков, LL-37 способствует апоптозу в эпителиальных и эндотелиальных клетках, а ROS, продуцируемые MPO, вызывают как апоптоз, так и некроз эпителиальных клеток [90]. Более того, внеклеточные гистоны (h4 и h5), высвобождаемые из NETs, участвуют в качестве основных эффекторов C5aR- и C5L2-опосредованного (рецепторы C5a) ALI у людей, крыс и мышей [91]. NET также обнаруживаются в моделях стерильного повреждения, таких как ALI, связанный с переливанием крови (TRALI) [92].NETs были связаны с ALI при гриппозном пневмоните, когда NET вызывали повреждение легких через ассоциацию с альвеолами в областях повреждения тканей [93]. NET обнаруживаются в мокроте пациентов с МВ [94]. Большая часть внеклеточной ДНК, обнаруженной в мокроте пациентов с МВ, на самом деле происходит от NET, поскольку комплексы ДНК соответствуют нейтрофильному ETosis и имеют сходную белковую сигнатуру [95]. Внеклеточная ДНК приводит к увеличению вязкости мокроты, что коррелирует с высокой концентрацией нейтрофилов и накоплением NET в дыхательных путях при МВ, что, следовательно, способствует микробной колонизации, пролиферации и образованию биопленок, вызывающих хроническое воспаление, коррелирующее с повышенной обструкцией легких [96, 97].Тем не менее, остается неясным, почему в дыхательных путях при МВ чаще встречается ЭТоз. Однако вполне вероятно, что NET образуются в ответ на бактерии-хозяева, такие как условно-патогенный микроорганизм Pseudomonas aeruginosa , один из основных патогенов, колонизирующих легкие при МВ, который также является распространенным патогеном, который, как известно, индуцирует NET [97]. И EET, и NET обнаруживаются в дыхательных путях пациентов с атопической астмой in vivo [98], тогда как NET, украшенные NE, гистоном h2 и цитруллинированным гистоном h4, обнаруживаются в мокроте пациентов с ХОБЛ [99, 100].Интересно, что генотипы Mycobacterium , M. tuberculosis (причина большинства типов туберкулеза) и M. canetti индуцировали образование NET и продукцию ROS в зависимости от времени [101]. НЕТ, индуцированные M. tuberculosis-, были украшены ключевыми этотическими маркерами, такими как гистон h3A, h3B и NE, и были способны улавливать, но не убивать M. tuberculosis [101]. Гранулемы являются важным и отличительным признаком туберкулеза и обычно вызываются микобактериальными или грибковыми инфекциями.Эти выступающие структуры представляют собой ключевой иммунный ответ на чужеродный материал, который слишком велик, чтобы его можно было очистить другими процессами иммунной защиты. Для более подробного обзора роли ЭТоза во время воспаления легких обратитесь к Cheng и Palaniyar [102]. Интересно, что, по-видимому, существует связь между активацией НАДФН-оксидазы, ЭТозом и апоптозом в иммунной защите от инфекционных агентов. Это было подчеркнуто исследованиями с участием нейтрофилов, полученных от пациентов с хронической гранулематозной болезнью (ХГБ; редкое наследственное заболевание НАДФН-оксидазы) и моделями ХГБ на мышах, где в обоих случаях ЭТотический ответ сильно снижен [76, 103].Кроме того, после фагоцитоза (in vitro) апоптоз нейтрофилов нарушается у больных ХГБ [104]. Неудачное разрешение воспаления у пациентов с ХГБ может привести к ряду воспалительных состояний легких, включая пневмонию, фиброз легких и абсцессы легких, и, в частности, у мышей с ХГБ, ОПН может быть результатом нарушения катаболизма триптофана (супероксид-зависимый процесс). [105].
Дополнительные процессы гибели клеток играют важную роль во время воспаления легких; к ним относятся аутофагия и некроптоз.Аутофагия влечет за собой внутриклеточную деградацию клеточных компонентов, которые затем доставляются в лизосомы для ферментативной деградации. Аутофагия может играть противоположную роль при хронических воспалительных заболеваниях легких и раке легких. Увеличение маркеров аутофагии, таких как образование аутофагосом, и уровней LC3B-II (ассоциированного с аутофагосомами белка) обнаруживается в легочном эпителии после индукции ALI у мышей после длительного воздействия гипероксии [106]. Во время туберкулеза аутофагия может способствовать генерации факторов антивирулентности [107], тогда как во время гриппа А аутофагия инфекции индуцируется репликацией вируса, зависящей от образования аутофагосом [108].Митофагия (избирательная деградация митохондрий посредством аутофагии) может в некоторых случаях усугубить тяжесть ХОБЛ, активируя дополнительные процессы гибели клеток, тогда как во время легочной гипертензии аутофагия может регулировать гибель клеток, облегчая защиту хозяина [106]. Более того, аутофагическая деградация и клиренс ресничек (цилиофагия) приводят к ассоциированной с ХОБЛ дисфункции ресничек [109]. Нарушение аутофагии может усугубить тяжесть муковисцидоза и идиопатического фиброза легких, а при раке легких — уменьшить канцерогенез; тем не менее, он также может способствовать выживанию опухолевых клеток.Следовательно, аутофагия может контролировать эффективность некоторых методов лечения рака [106]. Напротив, некроптоз (запрограммированный некроз), как известно, усиливает воспаление легких в нескольких моделях на мышах. В модели трансфузии эритроцитов и воспаления легких, индуцированного ЛПС, некроптоз эндотелиальных клеток легких индуцируется с помощью белка бокса 1 с высокой подвижностью (HMGB1) [110]. Staphylococcus aureus токсины могут вызывать некроптоз через рецептор-взаимодействующие протеинкиназы (RIP) 1 и 2, которые связываются с про-некротическим смешанным киназным геноподобным белком (MLKL) посредством передачи сигналов RIP1 / RIP2 / MLKL, что приводит к истощению альвеолярных макрофаги, а также экспрессия IL-1β, приводящая к повреждению легких [111].Некроптоз также наблюдался в эпителиальных клетках бронхов in vitro через индукцию сигаретным дымом, который также запускал высвобождение DAMP и провоспалительных цитокинов (IL-8, IL-6) [112]. In vivo сигаретный дым вызывает нейтрофильное воспаление дыхательных путей, о чем свидетельствует увеличение количества нейтрофилов, присутствующих в ЖБАЛ, которое было значительно снижено при лечении ингибитором некроптоза, некростатином-1 [112].
Эффероцитоз
Важнейшим процессом в успешном разрешении воспаления является эффективное удаление апоптотических клеток фагоцитами во время процесса, называемого эффероцитозом.Этот процесс помогает ограничить воспаление и поддерживать гомеостаз тканей. Эффероцитоз обеспечивает быстрое удаление апоптотических клеток до того, как они потеряют целостность мембраны и высвободят свое гистотоксичное внутриклеточное содержимое в окружающие ткани, что может вызвать повреждение тканей хозяина и усугубить воспаление. После поглощения апоптозные клетки содержатся в большой заполненной жидкостью пузырьке, называемой эфферосомой, которая сливается с лизосомами с образованием эфферолизосомы, которая в конечном итоге переваривает избыточную клетку.Эффероцитоз обычно осуществляется профессиональными фагоцитами, такими как макрофаги или дендритные клетки; однако этот процесс также может выполняться непрофессиональными фагоцитами, такими как эпителиальные клетки и фибробласты. Перед фагоцитозом апоптотические клетки претерпевают характерные морфологические изменения, такие как сокращение клеток, образование пузырей на мембранах и кариорексис, которые позволяют умирающей клетке распознаваться, поглощаться и впоследствии очищаться. В большинстве случаев фагоциты полностью поглощают умирающие клетки, например, макрофаги для очистки от апоптотических нейтрофилов [113].Однако в некоторых случаях, например, когда мишень слишком велика для эффективного фагоцитоза, несколько фагоцитов могут работать согласованно, поглощая апоптотические клетки частями «размером с укус». Такой сценарий наблюдается при эффероцитозе, осуществляемом фибробластами в отсутствие макрофагов [114].
Механизмы
Недавно рассмотренные Пун и др., Некоторые из ключевых механизмов процесса эффероцитоза включают высвобождение различных сигналов «найди меня» и «не дай мне», а также представление различных «съешь меня» и « сигналы «не ешь меня» от апоптотических клеток [10].Во время раннего апоптоза умирающие клетки привлекают фагоциты за счет высвобождения хемотаксических факторов. Эти сигналы find-me могут быть растворимыми или передаваться через субмикронные мембранные везикулы. Растворимые факторы включают нуклеотиды, которые высвобождаются из апоптотических клеток через мембранные каналы паннексина 1 (PANX1), активируемые каспазой [10]. Передача сигналов через субмикронные мембранные везикулы включает связанные с микрочастицами молекулы, такие как хемокиновый лиганд 1 CX3 (CX 3 CL1), ICAM3 и Ca 2+ -зависимый фосфолипид-связывающий белок аннексин A1 [10].Аннексин A1 высвобождается при нарушении целостности мембраны во время позднего апоптоза (вторичный некроз) и, как известно, способствует привлечению моноцитов через протеолитический процессинг белка 10, содержащего дисинтегрин и металлопротеиназный домен (ADAM10) [115], а также способствует апоптозу. поглощение и клиренс клеток [115, 116]. Интересно, что апоптотические клетки также обладают способностью сдерживать рекрутирование провоспалительных клеток посредством высвобождения запрещающих сигналов, которые действуют как негативные регуляторы миграции гранулоцитов.В настоящее время гликопротеин лактоферрин является единственным известным сигналом блокировки и высвобождается из различных типов апоптотических клеток, что впоследствии ингибирует миграцию нейтрофилов in vitro и in vivo [117] и миграцию эозинофилов in vitro [118]. Различные сигналы на поверхности клетки помогают фагоцитам отличать жизнеспособные клетки от апоптотических. Основным сигналом «съесть меня», экспонируемым на поверхности апоптотических клеток, является мембранный фосфолипид PS [66]. В жизнеспособных клетках PS ограничивается внутренней мембраной через трансмембранный транспортер липидов протеин-флиппазу.Однако во время ранней стадии апоптоза PS перемещается из внутреннего листка мембраны в наружный за счет активности фосфолипидной скрамблазы. PS, экспонированный на поверхности апоптотических клеток, может быть обнаружен фагоцитами с помощью нескольких механизмов распознавания. Прямое обнаружение PS происходит через различные мембранные рецепторы, включая специфический для мозга ингибитор ангиогенеза 1 (BAI1) [119], стабилизин-2 [120] и члены семейства белков Т-клеточного иммуноглобулинового домена (TIM), в частности TIM1, TIM3 и TIM4. [121, 122].Распознавание PS через BAI1 приводит к перестройке цитоскелета, чтобы способствовать поглощению фагоцитов, которое опосредуется через белок поглощения и клеточной подвижности 1 (ELMO1) -индикатор цитокинеза-180 (DOCK180) -Ras-связанный субстрат ботулинического токсина C3 ( RAC) (ELMO1-DOCK180-RAC) комплекс [119]. Стабилин-2 инициирует апоптотический захват клетками через связывание PS, опосредованное взаимодействиями с адаптерным белком поглощения (GULP) и тимозином β4 (регулирует полимеризацию актина) [123, 124], тогда как TIM4 преимущественно функционирует как связывающий белок для PS, где облегчается фагоцитарное поглощение. посредством передачи сигналов ассоциированных белков [125].Помимо этих подлинных мембранных рецепторов PS, PS также может связываться с помощью мостиковых молекул, включая фактор роста эндотелиального глобулы молочного жира 8 (MFG-E8), белок S и Gas6, которые являются лигандами, распознаваемыми их рецепторами клеточной поверхности на фагоцитах, в частности α v β 3 семейство рецепторов интегрина в случае MFG-E8 и семейство рецепторов Tyro3-Axl-Mer (ТАМ) в случае белка S и Gas6 [126–128]. Апоптотические клетки также могут экспонировать кальретикулин (резидентный белок 60 эндоплазматического ретикулума) на своей поверхности, что может служить дополнительным сигналом «съешь меня».Например, транслокация кальретикулина из эндоплазматического ретикулума в плазматическую мембрану происходит во время индукции апоптоза, сопровождаемого стрессом эндоплазматического ретикулума в раковых клетках, которые, в свою очередь, впоследствии очищаются фагоцитами посредством обнаружения кальретикулина CD91 (липопротеин-рецепторный белок низкой плотности). [129, 130]. В отличие от сигналов «есть меня», клетки также могут выставлять на своей клеточной поверхности сигналы «не есть меня». Это справедливо для жизнеспособных клеток, которые могут при определенных физиологических обстоятельствах перемещать PS в свой листок внешней мембраны.Однако эти жизнеспособные клетки избегают поглощения фагоцитами за счет воздействия сигналов «не ешьте меня», таких как CD31, CD47 и CD46 [10].
Регламент
Фагоцитарные функции могут быть усилены путем воздействия или лечения глюкокортикоидами (ГК). ГК также могут стимулировать макрофаги к переключению на противовоспалительный фенотип (M2), при котором они прекращают высвобождение провоспалительных цитокинов и одновременно высвобождают противовоспалительные цитокины (IL-10, TGFβ, IL-1ra), способствуя разрешению воспаление и восстановление тканей.ГК представляют собой класс кортикостероидов, которые представляют собой класс стероидных гормонов, регулярно используемых для лечения воспалительных заболеваний из-за их мощных противовоспалительных свойств. Кортизол является важным эндогенным GC, активно участвующим в модуляции различных метаболических, гомеостатических, иммунологических и сердечно-сосудистых функций. Однако при определенных хронических воспалительных состояниях эндогенного уровня кортизола недостаточно для подавления таких воспалительных поражений. В таких случаях синтетические (экзогенные) GC, такие как дексаметазон и гидрокортизон (которые могут быть более эффективными, чем эндогенные аналоги GC), могут быть введены для помощи и ускорения фазы разрешения воспаления.Что касается воспаления легких, ГК обычно используются для ограничения воспаления при заболеваниях легких, таких как астма и ALI / ARDS.
Макрофагальный эффероцитоз нейтрофилов усиливается в присутствии ГК, таких как дексаметазон и гидрокортизон, in vitro [131]. Макрофаги, обработанные дексаметазоном, также демонстрируют структурную реорганизацию цитоскелета и увеличение подвижности клеток, что важно для эффективного фагоцитоза [132]. Кроме того, дексаметазон увеличивал экспрессию активного RAC в макрофагах, ключевого сигнального белка, участвующего в различных клеточных функциях, включая фагоцитоз, а также подвижность клеток, митоз и заживление ран [132].ГК индуцируют белок S-зависимый эффероцитоз посредством передачи сигналов тирозинкиназы рецептора Mer, члена семейства тирозинкиназ рецептора ТАМ [133, 134]. ТАМ инициируют сигналы, которые регулируют клеточную функцию, а также определяют связывающую способность и фагоцитарный клиренс апоптотических клеток. У мышей с дефицитом ТАМ наблюдается нарушение способности к эффероцитозу, что связано с несколькими аутоиммунными заболеваниями. В очаге воспаления ГК могут стимулировать экспрессию Mer на фагоцитах [131]. Ингибирование Mer-опосредованного эффероцитоза у мышей усугубляет LPS-индуцированное повреждение легких, которое снижается за счет усиления передачи сигналов Mer посредством ингибитора TNFα-протеазы-0 (TAPI-0), специфического ингибитора расщепления Mer [135].Это подчеркивает, что Mer-опосредованный эффероцитоз является критическим процессом, который может модулировать патофизиологию легких. Было установлено, что протеолитическое расщепление от клеточной мембраны фагоцитов может снижать регуляцию Mer после воздействия провоспалительных стимулов, таких как LPS и блеомицин, при этом ингибирование этого протеолитического расщепления успешно достигается за счет блокады TAPI-0, которая впоследствии ингибирует подавление Mer и усиливает эффероцитоз у мышей, моделирующих индуцированное ЛПС и блеомицином повреждение легких [136, 137].Расшифровка молекулярных механизмов, лежащих в основе эффероцитоза и его регуляции посредством взаимодействия с ГК, поможет облегчить идентификацию новых терапевтических мишеней, способствующих разрешению воспаления и восстановлению тканей в легком, а также в других органах. Важно отметить, что эффективность ГК во время разрешения воспаления зависит от окружающей среды. ГК in vitro могут стимулировать апоптоз эозинофилов; однако известно, что они также задерживают апоптоз нейтрофилов.Тем не менее, во время гипоксии индуцируемые GC и провоспалительные цитокины эффекты выживания нейтрофилов теряются [138].
Другой способ, которым GCs, как полагают, проявляют и модулируют свои противовоспалительные способности, — это экспрессия и функция 37-кДа белка аннексина A1 (также известного как липокортин 1), нижестоящей эффекторной молекулы [139]. Аннексин А1 передает сигнал через рецептор, связанный с G-белком (GPCR), известный как рецептор формилпептида 2 (FPR2; ALXR у людей), который также является рецептором для биоактивного пролонгированного липидного липоксина A 4 [140].Аннексин A1 связывается с кислыми фосфолипидами мембран Ca 2+ -зависимым образом и экспрессируется в высоких уровнях в цитоплазме покоящихся клеток. В нейтрофилах человека> 60% цитоплазматического аннексина A1 хранится в гранулах желатиназы [141]. После активации клетки (например, в ответ на воспалительные стимулы) происходит быстрая транслокация аннексина A1 к листку внешней мембраны, где этот сигнал «найди меня» затем секретируется через различные клеточно-специфические молекулярные механизмы [139].Высвобождение эндогенного аннексина A1 из апоптотических нейтрофилов и макрофагов, обработанных GC (дексаметазоном), усиливает макрофагальный эффероцитоз нейтрофилов in vitro [142, 143]. Уровни экспрессии аннексина A1 из циркулирующих нейтрофилов и моноцитов повышаются у здоровых добровольцев после введения GC [144], при этом уровни экспрессии аннексина A1 также модулируются во время болезни. При болезни Кушинга (связанной с повышенным уровнем кортизола) лейкоциты обнаруживают заметно повышенный уровень внутриклеточного аннексина A1, а при болезни Аддисона (связанной с пониженным уровнем кортизола) лейкоциты демонстрируют заметно более низкие уровни внутриклеточного аннексина A1 по сравнению со здоровыми людьми [145 ].Врожденное высвобождение аннексина A1 иммунными клетками после лечения ГК может стимулировать апоптоз нейтрофилов и эффероцитоз макрофагов и ингибировать трансэндотелиальную миграцию нейтрофилов [139]. Данные in vivo, полученные на мышиной модели острого воспаления, подчеркивают, что аннексин A1 является ключевым регулятором во время естественного и вызванного GC разрешения воспаления [146]. Тем не менее, механизм регуляции аннексина A1 GC остается в значительной степени неясным. Между прочим, эффероцитоз также может быть усилен in vivo в легких мыши и в альвеолярных макрофагах от пациентов с ХОБЛ лечением статинами (препаратами, снижающими уровень холестерина), в частности лечением ловастатином, который усиливает эффероцитоз за счет ингибирования RhoA (регулятор цитоскелета) [147].
Успешное разрешение воспаления легких
Эффероцитоз, осуществляемый резидентными фагоцитами легких, определяет успешное разрешение воспаления легких и регулирует нормальную структуру легких. Профессиональные фагоциты включают альвеолярные макрофаги, интерстициальные макрофаги легких и дендритные клетки легких, тогда как непрофессиональные фагоциты включают эпителиальные клетки легких, такие как альвеолярные и бронхиальные эпителиальные клетки. Дефектный эффероцитоз, который приводит к увеличению количества апоптотических клеток, вовлечен в ряд заболеваний легких, включая астму, ALI, CF и COPD, и хорошо рассмотрен [148, 149].Кроме того, высокоспециализированные биоактивные липиды играют ключевую роль на этапе разрешения воспаления.
Профессиональные фагоциты легких
Альвеолярные макрофаги составляют самую многочисленную популяцию профессиональных фагоцитов в альвеолярном пространстве, где они могут составлять 90–95% популяции клеток из здоровой жидкости БАЛ. Эти фагоциты обладают огромной фаголизосомальной способностью убивать проглоченные микробы. Апоптоз альвеолярных макрофагов, которые проглотили Streptococcus pneumonia , необходим для уничтожения и удаления этих бактерий, что способствует разрешению на мышиной модели легочной инфекции [150, 151].Хотя альвеолярные макрофаги способны фагоцитировать разнообразные повреждающие агенты, в состоянии покоя скорость эффероцитоза альвеолярных макрофагов несколько ниже, чем у других тканевых макрофагов [152]. Дефекты эффероцитоза альвеолярных макрофагов объясняются несколькими факторами, включая снижение адгезии и активацию через SP-A и SP-D (эффективные регуляторы функции макрофагов) трансмембранного рецепторного сигнала, ингибирующего регуляторный белок альфа (SIRPα) [153]. Однако эти дефекты эффероцитоза преодолеваются за счет рекрутирования мононуклеарных фагоцитов во время острых воспалительных сценариев, таких как острое воспаление легких [153].В отличие от циркулирующих моноцитов, дендритных клеток и тканевых макрофагов, альвеолярные макрофаги обладают многочисленными рецепторами распознавания апоптотических клеток, что означает, что эти клетки чрезвычайно чувствительны к апоптотической гибели клеток в альвеолярном пространстве [154]. Альвеолярные макрофаги в высокой степени экспрессируют все три рецептора ТАМ, при этом показано, что блокада этих рецепторов дополнительно подавляет эффероцитоз, но не фагоцитарное связывание [152]. В легких также может находиться повышенное количество интерстициальных макрофагов, что обычно наблюдается у курильщиков и пациентов с ХОБЛ.Хотя способность интерстициальных макрофагов легких к эффероцитозу остается в значительной степени неясной, они, по-видимому, играют ключевую роль в патогенезе эмфиземы, вызванной сигаретным дымом, у мышей посредством высвобождения TNF и IL-6 [155]. В легком есть два подмножества дендритных клеток, и в легком мыши было продемонстрировано, что подмножество дендритных клеток CD103 + способствует эффероцитозу и представляет антигены, ассоциированные с апоптотическими клетками, Т-клеткам CD8 + [156] .Подобно способности интерстициальных легочных макрофагов к эффероцитозу, существует также мало исследований способности дендритных клеток легких человека к эффероцитозу, что является областью, которая также заслуживает большего внимания.
Непрофессиональные фагоциты легких
Сообщалось о распознавании и последующем эффероцитозе эозинофилов бронхиальными и альвеолярными эпителиальными клетками человека in vitro, которые были усилены лечением дексаметазоном [157, 158]. Распознавание апоптозных эозинофилов оказалось как лектин-, так и интегрин-зависимым [157] с поглощением апоптотических клеток с участием αvβ3-, CD36-, αvβ5- и событий, опосредованных рецепторами PS [158].Таким образом, эти данные предполагают непассивную роль эпителия дыхательных путей во время эозинофильного доминантного воспаления при астме. Совсем недавно на трансгенных мышах с моделями аллергического воспаления дыхательных путей группа Равичандрана смогла продемонстрировать RAC1-зависимый эффероцитоз эпителиальных клеток дыхательных путей эпителиальными клетками бронхов, что привело к высвобождению противовоспалительных цитокинов [159] (см. Таблицу 1 для обзор противовоспалительных цитокинов и связанных с ними биоактивностей). Между прочим, индуцируемая делеция экспрессии RAC1 в эпителиальных клетках дыхательных путей мыши приводит к нарушению эффероцитоза и генерации атипичного профиля противовоспалительных цитокинов в эпителиальных клетках бронхов [159].Тем не менее, в настоящее время остается явный недостаток исследований, пытающихся манипулировать возможностями эффероцитоза в эпителиальных клетках легких в современных моделях воспаления дыхательных путей. Содействие таким исследованиям может помочь раскрыть новые терапевтические подходы к лечению различных воспалительных заболеваний легких.
Таблица 1 Противовоспалительная биоактивность различных цитокинов
Про-разрешающие биоактивные липиды
Зависимое от липоксигеназы (LOX) ферментативное превращение субстратов полиненасыщенных жирных кислот (PUFA) в медиаторы, производные PUFA, является еще одним важным биохимическим процессом, который является ключом к модуляции воспаления [5, 160].ПНЖК высвобождаются из клеточных мембран активированных клеток, таких как нейтрофилы, моноциты / макрофаги, лимфоциты и эозинофилы, затем ферментативно превращаются в специализированные липидные медиаторы, способствующие разрешению, которые различаются по структуре и функциям [161]. Эти пролонгированные липидные медиаторы включают липоксины (A 4 и B 4 ), резольвины D- и E-серий, протектины и марезины, и они активны от пикограмм до нанограмм. Эти биоактивные липиды часто обладают двойной противовоспалительной биоактивностью и биоактивностью, способствующей разрешению (обзор см. В таблице 2), которые превосходно рассмотрены группой Серхана [21, 162, 163].В клинике используются фармакологические ингибиторы, в том числе те, которые нацелены на определенные липоксигеназы, из-за их способности подавлять побочные эффекты, сопровождающие воспаление; однако эти ингибиторы также могут нарушать эндогенное производство других биоактивных липидов [164–166]. Напротив, аспирин и глюкокортикоид дексаметазон могут инициировать эндогенные противовоспалительные пути посредством активации рецептора липоксина A 4 AXLR / рецептора формилпептида, подобного рецептору-1 (FPRL1) [140], GPCR, который теперь называется AXLR / FPR2 [ 167].Эти способствующие рассасыванию биоактивные липиды участвуют в ряде воспалительных заболеваний легких, включая астму, муковисцидоз, интерстициальное заболевание легких, респираторное заболевание, обостренное аспирином, ХОБЛ и эмфизему [161].
Таблица 2 Специализированные биоактивные липиды, способствующие разрешению воспаления
Липоксины и резолвины
В очагах воспаления ПНЖК арахидоновая кислота (ADA) может метаболизироваться до простагландинов и лейкотриенов (таких как LTB 4 ), но она также превращается в семейство про-рассасывающихся биоактивных липидов, называемых липоксинами. , который может подавлять воспаление, вызванное лейкотриенами [168].В настоящее время липоксины являются наиболее изученным семейством липидов, способствующих разрешению воспаления, и их уровни значительно повышаются во время фазы разрешения воспаления [5]. Во время воспаления дыхательных путей ферментативный гидролиз посредством активности нейтрофилов 5-LOX и эпителиальной 15-LOX приводит к биосинтезу липоксина A 4 и B 4 [4]. Дополнительный биосинтез липоксина A 4 и B 4 может происходить через взаимодействия тромбоцитов и нейтрофилов (через тромбоциты 12-LOX), например, в сосудистой сети [169], а также различными типами клеток, включая нейтрофилы, эозинофилы и альвеолярные макрофаги, хотя и в меньшей степени [5, 169, 170].Помимо ADA, в местах воспаления присутствуют дополнительные PUFA, такие как докозагексаеновая кислота (DHA) и эйкозапентаеновая кислота (EPA), которые могут ферментативно превращаться в резольвины D- и E-серий соответственно. Во время воспаления сосудов DHA преобразуется ацетилированной аспирином эндотелиальной клеткой циклооксигеназой-2 (ЦОГ-2) через активность нейтрофилов LOX, что приводит к гидролизу резольвинов D-серии [161]. При аналогичном воспалительном сценарии EPA превращается в резольвины E-серии под действием COX-2, происходящего из ацетилированных аспирином эндотелиальных клеток, где биосинтез резольвинов E-серии включает прямую трансформацию нестабильных промежуточных продуктов EPA активированными лейкоцитами [161].GPCR AXLR / FPR2 служит рецептором для липоксина A 4 и резольвина D1 [171, 172], которые также могут быть активированы аннексином A1 через индукцию глюкокортикоидов [140]. Люди экспрессируют ALXR / FPR2 в лейкоцитах и тканевых резидентных клетках [171], причем экспрессия рецепторов модулируется местными медиаторами воспаления. Резолвин D1 также связывается с другим GPCR, а именно с GPR32, который также известен как рецептор резолвина D1 (DRV1) и экспрессируется лейкоцитами [173]. Однако из-за отсутствия мобилизации GPR32 из нейтрофилов человека активность резольвина D1 по разрешению в первую очередь обеспечивается путями передачи сигналов ALXR / FPR2, которые подтвердили, что у мышей отсутствует рецептор ALXR / FPR2 [174].Резолвин E1 может связываться с дополнительными GPCR, а именно с рецептором хемокиноподобного 1 (CMKLR1) и рецептором 1 LTB 4 (BLT1), экспрессируемым полиморфноядерными (PMN) лейкоцитами [175].
Протектины и марезины
Протектины образуются 15-LOX-зависимым образом, который катализирует превращение DHA в протектин D1 через промежуточный эпоксид [176]. Кроме того, аспирин может инициировать биосинтез протектина из DHA посредством ацетилирования ЦОГ-2 [177]. Известно, что пролонгированное биологическое действие протектина D1 является клеточно-специфическим; следовательно, вполне вероятно, что такие взаимодействия опосредуются одним или несколькими специфическими рецепторами.Однако рецепторы, специфичные для протектина 1, еще предстоит идентифицировать [161]. Несколько позже были открыты марезины (полученные из макрофагов), которые представляют собой еще одно семейство про-разрешающих биоактивных липидов [178]. Подобно протектинам и резольвинам серии D, марезины также являются производными DHA. Однако биосинтез марезина 1 происходит с помощью нового промежуточного эпоксида, который также усиливает превращение макрофагов с фенотипа M1 в M2, причем макрофаги M2 способны продуцировать повышенные уровни этого пролегающего липидного медиатора из промежуточного эпоксида по сравнению с макрофагами M1. [179].Важно отметить, что с точки зрения прорешения, марезин 1 также способен увеличивать эффероцитоз макрофагов и способствовать регенерации тканей [178, 180]. Идентификация рецепторов, специфичных для марезина 1, еще предстоит; однако очевидно, что участвуют специфические GPCR [180].
Биоактивные липиды при воспалении легких
Нарушение образования этих пролонгированных липидных медиаторов во время воспаления дыхательных путей может привести к хроническим воспалительным заболеваниям легких. Снижение образования липоксина описано при тяжелой / неконтролируемой астме, муковисцидозе, респираторных заболеваниях, обостренных аспирином, и интерстициальной склеродермии [181–184].Во время тяжелой / неконтролируемой астмы дисрегулируемая экспрессия генов биосинтеза липоксинов частично ответственна за снижение продукции липоксинов (см. Обзор Levy et al. [185]). Повышенные уровни DHA обнаруживаются в слизистой оболочке дыхательных путей у здоровых людей; однако при астме и муковисцидозе уровни DHA в слизистой оболочке снижаются [186]. Резолвины E1 и D1 могут улучшать разрешение аллергического воспаления дыхательных путей на мышиных моделях астмы [187, 188]. Резолвин E1 уменьшал количество макрофагов, эозинофилов и лимфоцитов, присутствующих в БАЛ, а также улучшал гиперреактивность дыхательных путей и метаплазию слизи дыхательных путей после ингаляции метахолина у мышей [189].В частности, резолвин E1 улучшает разрешение аллергического воспаления дыхательных путей за счет снижения выработки IL-6, IL-17 и IL-23 в легких мыши, с повышением разрешения за счет увеличения выработки IFNγ и липоксина A 4 в легких. мышей, получавших резолвин E1 [189]. Недавно резолвин D4 был идентифицирован в тканях человека и подтвержден его сильнодействующий про-разрешающий эффект при инфекциях, вызванных S. aureus у мышей [190, 191]. Однако точную роль резолвина D4 при воспалении легких еще предстоит установить.Подобно уровням липоксина при тяжелой / неконтролируемой астме, снижение уровня протектина D1 также наблюдается во время обострений астмы [192]. Протектин D1 может также снижать аллергические реакции дыхательных путей на мышиной модели астмы, где внутривенное введение протектина D1 (до провокации аэроаллергеном) приводит к ослаблению инфильтрации эозинофилов и высвобождению провоспалительных цитокинов [192]. Кроме того, после посталлергенного заражения (после того, как легочное воспаление было установлено), протектин D1 способен проявлять свойства, способствующие разрешению, которые ускоряют разрешение аллергического воспаления дыхательных путей [192].Взятые вместе, приведенные выше данные указывают на то, что эти биоактивные липиды являются ключевыми эффекторами цепей, способствующих разрешению во время воспаления легких, и что нарушение их эндогенных уровней способствует нескольким воспалительным заболеваниям легких. Для подробного обзора роли, которую играют биоактивные липиды, способствующие разрешению, во время воспаления легких, см. [161].
Нарушение регуляции / нарушение разрешения воспаления легких
Воспаление с преобладанием нейтрофилов
Поскольку наиболее распространенные клетки и во многих отношениях самые нечеткие инструменты иммунного арсенала, вероятно, неудивительно, что нейтрофильное воспаление является признаком многочисленных воспалительных состояний легких .Более того, легкие являются основными очагами воспаления и травм, поскольку нейтрофилы сохраняются в легких намного дольше, чем в других органах [195]. Первые респонденты как на эндогенные, так и на экзогенные раздражители, их роль в острых расстройствах не вызывает сомнений, но они также участвуют в патогенезе множества хронических состояний, что свидетельствует о сбое нормальных механизмов, с помощью которых происходит разрешение. Ниже мы используем несколько демонстрационных условий, чтобы проиллюстрировать механизмы нейтрофильного воспаления, разрешения и развития хронической болезни.
Пневмония
Пневмония, являющаяся значительным бременем заболеваемости и смертности как в Великобритании, так и во всем мире, представляет собой острую воспалительную реакцию на инфекцию нижних дыхательных путей, которая видна на рентгеновском снимке грудной клетки. Это происходит в ответ на действие различных патогенов, чаще всего бактерий и вирусов, и обычно запускается при распознавании консервативных «чужеродных» рецепторов. Когда ответ хозяина успешен в сдерживании и поглощении ответственного патогена, инфекция остается локализованной, и легкое может зажить без последствий.Острый сбой этого процесса приводит к диссеминированной инфекции, которая в конечном итоге может привести к смерти. В среднесрочной и долгосрочной перспективе неэффективность секвестрации и разрешения предрасполагает к эмпиеме, формированию абсцесса и бронхоэктазии. Несмотря на лучшее понимание основных механизмов воспаления и травм, основой лечения пневмонии остаются противомикробные препараты. К сожалению, остается подгруппа пациентов, у которых даже комбинация защиты хозяина и антибиотиков не справляется с инфекцией и у которых развивается полиорганная недостаточность (см. Ниже).В последние годы проявился больший интерес к изучению роли модуляции иммунного ответа при тяжелой пневмонии, и множество различных препаратов продемонстрировали некоторый потенциал в улучшении результатов.
Streptococcus pneumoniae является наиболее частым возбудителем внебольничной пневмонии (ВП) и может вызывать различные степени тяжести заболевания. Стрептококковая инфекция связана с плотным нейтрофильным воспалением и активацией каскада свертывания через рецептор тромбина, рецептор 1, активируемый протеиназой (PAR-1).Было разработано множество антагонистов PAR-1, и модели на животных предполагают, что подавление активации коагуляции посредством этого пути может снизить нагрузку на дыхательные пути нейтрофилами, продукцию воспалительных цитокинов и альвеолярную утечку без ущерба для бактериального клиренса [196]. Кроме того, доказательства основных исследований показывают, что существующие антитромбоцитарные агенты, хорошо зарекомендовавшие себя при лечении сердечно-сосудистых заболеваний, могут еще улучшить исходы при пневмонии за счет снижения активации каскада свертывания крови и, таким образом, прогрессирования до ALI и ARDS [197].Активированный протеин C (APC) представляет собой эндогенный противовоспалительный и антикоагулянтный хемокин, который участвует в предотвращении диссеминированной инфекции и контроле источников. Недавно было подчеркнуто, что на животных моделях стрептококковой пневмонии сверхэкспрессия APC снижает распространение бактерий на другие органы и нейтрофильное воспаление в очаге первичной инфекции [198]. Наряду с исследованием путей, участвующих в острой воспалительной реакции, растет интерес к лекарствам, играющим роль в лечении хронических заболеваний легких, таким как макролиды.Макролиды давно пользуются популярностью, поскольку они проявляют как антибактериальные, так и противовоспалительные свойства и полезны в условиях, когда бактериальная колонизация является признаком, включая диффузный панбронхиолит, муковисцидоз и бронхоэктазы. Недавнее пилотное исследование выявило тенденцию к снижению уровней циркулирующих провоспалительных цитокинов у пациентов с ВП, получавших макролиды, по сравнению с пациентами, получавшими другие классы антимикробных препаратов, особенно через 5-7 дней после инфицирования [199].В последнее десятилетие было все более широко признано, что ингибиторы HMG Co-A редуктазы (статины) не только снижают липидную нагрузку и модифицируют сердечно-сосудистые заболевания, но и обладают недооцененными противовоспалительными эффектами, которые потенциально можно использовать для лечения воспалительных заболеваний. Недавнее исследование показало, что аторвастатин снижает тяжесть кашля при стабильных бронхоэктазиях с ассоциированным увеличением апоптозных нейтрофилов, обнаруживаемых в мокроте, и возобновился интерес к их роли в модуляции острых воспалительных состояний [200].В недавнем систематическом обзоре был изучен ряд исследований, изучающих роль статинов в ВП, и сделан вывод, что они модулируют нейтрофильный ответ, снижают нагрузку циркулирующих цитокинов и потенциально влияют на смертность [201].
Продолжительность жизни нейтрофилов является ключевым фактором воспалительного ответа и очевидной мишенью для поиска новых противовоспалительных средств. В условиях инфекции проблема заключается в безопасном истощении количества нейтрофилов до уровня, который может облегчить краткосрочные и долгосрочные заболевания без ущерба для защиты хозяина и предрасположенности к системной инфекции.Имеются данные о том, что при многочисленных острых и хронических заболеваниях легких, включая ВП, неспособность своевременно развить апоптоз нейтрофилов способствует развитию заболеваемости [202]. Ингибиторы циклинзависимых киназ (CDKis) — это группа препаратов, которые активно исследуются на предмет их способности останавливать развитие клеточного цикла и индуцировать апоптоз даже в терминально дифференцированных клетках, таких как нейтрофилы, путем подавления Mcl-1, и в настоящее время проводится несколько исследований. которые продемонстрировали свой потенциал в качестве безопасных и эффективных модуляторов воспаления [203–205].
ARDS
Синдром мультиорганной дисфункции (MODS) — часто смертельное состояние, вызванное сильнейшим воспалительным инсультом, которое приводит к парадоксально бесполезной агрессивной реакции слизистой оболочки. ОРДС является респираторным компонентом этого заболевания и возникает в результате патологической реакции, известной как диффузное альвеолярное повреждение, которое возникает вторично по отношению к безудержному нейтрофильному воспалению [206]. Существует множество причин ОРДС, включая сепсис, шок, травму и желудочную аспирацию [207, 208].Прогрессирование до ОРДС является маркером тяжелого сепсиса и связано с плохими результатами. Терапия СПОН остается в значительной степени поддерживающей, но растет интерес к роли иммунной модуляции как дополнения к антибиотической или другой терапии для ослабления гиперактивного иммунного ответа, что в конечном итоге приводит к серьезному нарушению газообмена и дыхательной недостаточности. Смертность при ОРДС коррелирует с числом нейтрофилов и уровнями циркулирующих провоспалительных цитокинов, предполагая, что использование воспалительной реакции может быть ключом к улучшению результатов [209, 210].
Несмотря на продолжающиеся споры, глюкокортикоиды остаются наиболее изученной противовоспалительной стратегией при ОРДС. Имеются некоторые данные, позволяющие предположить, что при раннем течении болезни внутривенные стероиды снижают потребность в механической вентиляции легких, продолжительность пребывания в ITU и улучшают оксигенацию с умеренным влиянием на смертность [211–214]. Однако такой успех может перевесить потенциальные осложнения только в условиях бдительного наблюдения за нозокомиальной инфекцией и отказа от нервно-мышечной блокады из-за возможных осложнений при лечении стероидами.Более того, было высказано предположение, что если оставить его на более поздний срок, то есть> 14 дней после начала заболевания, введение стероидов может вызвать парадоксальное увеличение смертности [211]. Следовательно, терапия глюкокортикоидами еще не доказана как эффективная терапия ОРДС и не рекомендуется для лечения, если не известно, что она является вторичной по отношению к стероид-чувствительному инсульту. Ранние исследования на животных моделях показывают, что макролидные антибиотики могут продемонстрировать эффективность в лечении ОРДС [215, 216]. Хотя это подтверждается обсервационным исследованием, которое подчеркивает тенденцию к снижению смертности при лечении макролидами, доказательства остаются недостаточно надежными, чтобы поддерживать их использование в качестве рутинного метода лечения [217, 218].Возможно, почти столь же поучительно, как и многообещающие методы лечения, которые, несмотря на хорошее биохимическое обоснование, не оказались клинически эффективными. Несмотря на известную роль метаболитов, производных циклооксигеназы, в сепсисе и его последствиях, исторически было продемонстрировано, что использование нестероидных препаратов не приводит к снижению частоты ОРДС, ассоциированного с сепсисом [219].
Муковисцидоз
CF является наиболее распространенным аутосомно-рецессивным заболеванием, ограничивающим жизнь, у кавказцев с частотой 1 случай на 2500.Вызванное мутациями потери функции муковисцидоза, трансмембранного регулятора проводимости (CFTR), эпителиального хлоридного канала, это гетерогенное мультисистемное воспалительное заболевание, основными клиническими проявлениями которого являются тяжелые прогрессирующие бронхоэктазы и внешнесекреторная недостаточность поджелудочной железы. Классическим патофизиологическим объяснением заболевания легких при МВ является «гипотеза малого объема», согласно которой аномальные электрохимические градиенты поверхности дыхательных путей, вторичные по отношению к потере CFTR, приводят к увеличению поглощения воды и внеклеточных катионов [220].Возникающее в результате обезвоживание жидкости на поверхности дыхательных путей способствует гиперсекреции слизи, подавляет мукоцилиарный клиренс и выводит из строя катионные защитные пептиды хозяина, что приводит к непрерывным циклам синопульмональных инфекций, которые в конечном итоге прогрессируют до хронического воспаления с ремоделированием дыхательных путей. После развития структурного заболевания легких при МВ он редко регрессирует, и, следовательно, существует острая необходимость в разработке эффективных вариантов раннего лечения для снижения долгосрочной заболеваемости [221].
В последние годы были предприняты попытки изучить роль аберрантной иммунной функции у пациентов с МВ, поскольку очевидно, что существуют детерминанты тяжести заболевания (легких), не связанные с CFTR, и многочисленные намеки на аномальные воспалительные реакции.Необычная восприимчивость пациентов с МВ к патогенам «низкой вирулентности», таким как P. aeruginosa и Burkholderia cepacia , повышенная частота аллергических заболеваний дыхательных путей и растущие свидетельства «энтеропатии, связанной с МВ» и системного воспаления: назовите лишь несколько [222–224]. Несмотря на десятилетия исследований, до недавнего времени было очень мало прогресса в разработке новых вариантов лечения CF, хотя многообещающие препараты, непосредственно восстанавливающие функцию CFTR, недавно стали доступны для небольшой группы пациентов [225].Для остальной части популяции CF, возможно, лучшая надежда заключается в разработке безопасных и эффективных противовоспалительных средств, которые можно использовать синергетически с антибиотиками для предотвращения хронического воспаления и повреждения дыхательных путей.
Ряд групп продемонстрировали внутреннюю недостаточность врожденной иммунной системы при МВ, и продолжаются дискуссии о том, является ли это результатом недооцененной до сих пор роли CFTR в функции нейтрофилов или вторичным по отношению к хронической воспалительной среде у взрослых пациентов с МВ.Отсроченный апоптоз нейтрофилов, аберрантная фаголизосомная деструкция Pseudomonas и избыточная продукция IL-8 описаны у пациентов с МВ и предлагают множество терапевтических целей [223, 226, 227]. Как обсуждалось выше, есть большая надежда, что CDKis могут предложить новый подход к иммунной модуляции при воспалительных расстройствах, поскольку их роль в лечении доброкачественных расстройств изучается. Имеются предварительные данные о том, что CDKis может корректировать отсроченный апоптоз нейтрофилов МВ, что дает надежду на коррекцию воспалительного ответа и, возможно, повышение эффективности антимикробных препаратов в потенциале клиренса установленных патогенов дыхательных путей [227].Действительно, с разработкой новых водорастворимых ингибиторов CDK, возможность распыления терапии остается открытой и может снизить системные эффекты при одновременном повышении эффективности. Помимо парадоксальной неспособности удалить патогены, сохранение нейтрофилов в дыхательных путях при МВ приводит к избытку протеаз, производных от PMN, которые не только напрямую повреждают респираторный эпителий, но также снижают эффективность фагоцитарного клиренса [228, 229].
Традиционные противовоспалительные стратегии исторически апробировались у пациентов с МВ, но по-прежнему ограничены значительными побочными эффектами и остаются нереализованными в повседневной клинической практике [230, 231].Роль «местных» традиционных противовоспалительных средств, таких как небулайзированные нестероидные противовоспалительные препараты (НПВП), мало изучена, хотя этот путь может предложить безопасную и более эффективную возможность облегчить воспаление легких при минимизации системных эффектов [ 232]. Ингаляционные кортикостероиды обычно предназначены для пациентов с сопутствующей гиперреактивностью дыхательных путей, поскольку они демонстрируют более чувствительный к стероидам воспалительный инфильтрат дыхательных путей. Несколько групп продемонстрировали повышенные уровни LTB 4 в дыхательных путях МВ, и был достигнут некоторый успех в использовании монтелукаста, антагониста лейкотриеновых рецепторов, хорошо зарекомендовавшего себя при лечении астмы, для уменьшения респираторных симптомов у пациентов с МВ [233, 234].Возможно, неудивительно, учитывая распространенность микробной колонизации, макролиды имеют долгую историю болезни легких. Принято считать, что их роль связана как с их противовоспалительными свойствами, так и с отсроченным антимикробным действием, что снижает количество патогенов [235–238]. К сожалению, в последнее время появились данные, позволяющие предположить, что длительное использование макролидов связано с увеличением частоты мультирезистентных атипичных микобактериальных инфекций у пациентов с МВ, что в конечном итоге может ограничить их использование [239].Одной из проблем, связанных с разработкой нового агента, является длительный период времени от рабочего стола до постели больного, и поэтому существует большой интерес к изучению альтернативных вариантов использования лекарств с хорошо установленными профилями безопасности и переносимости. Статины, давно известные своей ролью в модуляции липидов в сыворотке крови, теперь, как известно, обладают более широкими противовоспалительными свойствами, и потенциальные последствия этого в настоящее время изучаются в различных условиях. Известно, что IL-8 содержится в сыворотке крови при МВ, и недавно было продемонстрировано, что флувастатин способен снижать уровень IL-8 и в конечном итоге может помочь подавить системное воспаление [240].
Воспаление с преобладанием эозинофилов (астма)
Эозинофилы важны при аллергическом воспалении дыхательных путей. Комбинированные повреждающие эффекты, возникающие в результате большого количества инфильтрирующих эозинофилов, замедленного апоптоза эозинофилов и нарушения эффероцитоза, могут вызывать хроническое воспалительное заболевание легких, такое как астма. Астма — это спектр состояний, определяемых общей патологией обратимой обструкции дыхательных путей и гиперчувствительностью слизистой оболочки дыхательных путей к антигенам окружающей среды.Чаще всего это происходит как часть аллергического синдрома атопических расстройств, хотя может возникать и изолированно. У пациентов с астмой развивается чувствительность к антигенам окружающей среды, таким как перхоть животных и растительный материал, и при воздействии этих антигенов развивается аллергическая реакция 1 типа, приводящая к бронхоспазму, хрипу, кашлю и гиперсекреции слизи, что приводит к ограничению воздушного потока.
У примерно 10% взрослого населения Великобритании астма является распространенным заболеванием многофакторного происхождения, и исследования показывают, что и генетика, и окружающая среда играют значительную роль.У предрасположенных людей «нормальные» антигены окружающей среды мигрируют через эпителий дыхательных путей и представляются наивным Т-клеткам, которые запускают активацию продукции IgE В-клетками. IgE взаимодействует с рецепторами на поверхности резидентных тучных клеток ткани, и дальнейшее воздействие антигена приводит к перекрестному связыванию IgE с активацией клеток. Результирующая дегрануляция тучных клеток вызывает высвобождение медиаторов, включая гистамин, LTB 4 , IL-8, IL-10 и TNF, вызывая острую воспалительную реакцию.Поздняя фаза астматического ответа обычно возникает через 6–9 часов после воздействия антигена и возникает вторично по отношению к постоянной секреции цитокинов, например IL-5, GM-CSF и IL-3, которые способствуют миграции эозинофилов, устойчивости и долголетию в легких и образуют основу стойкого воспаления дыхательных путей у пациентов с астмой.
Основой лечения астмы являются глюкокортикоиды, обычно вводимые в виде ингаляционных препаратов, которые притупляют воспалительную реакцию и вызывают апоптоз эозинофилов.Эта терапия с дополнительными бронходилататорами достаточна для контроля симптомов у большинства пациентов, но не обладает тонкостью и значительно ограничена токсичностью. Возможно, неудивительно, что остается подгруппа пациентов, которые не реагируют на этот подход и имеют постоянно неконтролируемые симптомы, которые в конечном итоге могут привести к ремоделированию дыхательных путей. Следовательно, существует потребность в новых специфических ингибиторах эозинофильного воспаления в легких, которые могут в достаточной мере контролировать симптомы и проявлять минимальные системные токсические эффекты [241].Использование дополнительных антагонистов лейкотриеновых рецепторов для лечения умеренно-тяжелой астмы, которые избирательно подавляют провоспалительные эффекты лейкотриенов, показало хорошие результаты, что подчеркивает правильность принципа таргетной терапии [242].
Отсроченный апоптоз эозинофилов и, как следствие, сохранение в дыхательных путях остается основным патологическим признаком астмы и одной из целей стероидной терапии. Недавние исследования in vitro и на мышах продемонстрировали, что, как и в случае нейтрофилов, ингибиторы CDK способны вызывать апоптоз как циркулирующих, так и воспалительных эозинофилов посредством подавления Mcl-1, хотя значение этого в клинических условиях остается неясным [243–245].Недавнее исследование, посвященное изучению новых модуляторов апоптоза эозинофилов, показало, что перекись водорода вызывает гибель клеток и ускоряет разрешение воспаления дыхательных путей каспазозависимым образом, а также ускоряет восстановление функции легких [246]. Как обсуждалось ранее, в последние годы большое внимание уделялось роли эндогенных липидных медиаторов разрешения, например липоксины, резольвины и протектины, а также потенциальную роль, которую они могут играть в ослаблении вредной реакции при воспалительных состояниях [69].Резолвин D1, один из таких медиаторов, и его аналог, вызываемый аспирином, резолвин D1, как было показано, значительно снижают эозинофилию дыхательных путей и гиперсекрецию слизи за счет уменьшения деградации IL-5 [188]. Сообщалось, что липоксин A 4 подавляет ответы эозинофилов посредством подавления активации GM-CSF [247]. Его функционально связанный, хотя структурно отличный аналог липоксина B 4 , способствует разрешению аллергических реакций в верхних и нижних дыхательных путях за счет снижения хемотаксиса эозинофилов и дегрануляции тучных клеток, подчеркивая потенциал терапевтического использования этих путей при эозинофильных нарушениях дыхательных путей [248].